Int. J. Dev. Biol. 66: 391 - 400 (2022)
Single-cell transcriptomics defines Dot1L interacting partners and downstream target genes in the mouse molar dental pulp
Original Article | Published: 21 February 2023
Abstract
Although histone methyltransferases are implicated in many key developmental processes, the contribution of individual chromatin modifiers in dental tissues is not well understood. Using single-cell RNA sequencing, we examined the expression profiles of the disruptor of telomeric silencing 1-like (Dot1L) gene in the postnatal day 5 mouse molar dental pulp. Dot1L is the only known enzyme that methylates histone 3 on lysine 79, a modification associated with gene expression. Our research revealed 15 distinct clusters representing different populations of mesenchymal stromal cells (MSCs), immune cells, pericytes, ameloblasts and endothelial cells. We documented heterogeneity in gene expression across different subpopulations of MSCs, a good indicator that these stromal progenitors undergo different phases of osteogenic differentiation. Interestingly, although Dot1L was broadly expressed across all cell clusters within the molar dental pulp, our analyses indicated specific enrichment of Dot1L within two clusters of MSCs, as well as cell clusters characterized as ameloblasts and endothelial cells. Moreover, we detected Dot1L co-expression with protein interactors involved in epigenetic activation such as Setd2, Sirt1, Brd4, Isw1, Bptf and Suv39h1. In addition, Dot1L was co-expressed with Eed2, Cbx3 and Dnmt1, which encode epigenetic factors associated with gene silencing and heterochromatin formation. Dot1l was co-expressed with downstream targets of the insulin growth factor and WNT signaling pathways, as well as genes involved in cell cycle progression. Collectively, our results suggest that Dot1L may play key roles in orchestrating lineage-specific gene expression during MSC differentiation.
Keywords
Dot1L, histone methyltransferase, dental pulp, stem cells, H3K79me2
Introduction
The dental pulp is the neurovascular bundle central to each tooth, and consists of nerve fibers, blood vessels and stromal cells (Sui et al., 2019). The dental pulp is composed of a heterogenous population of MSCs exhibiting self-renewal, a high capacity for multi-lineage differentiation, and the potential to regenerate a dentin/pulp-like complex (Tsutsui, 2020). The fate of the dental pulp cells is regulated by multiple transcription factors (e.g., Msx1, Lef1, Pitx2 and Runx2) and growth factors (e.g., basic fibroblast growth factor, transforming growth factor-β and bone morphogenic proteins) (Zhang et al., 2005). However, an important issue that remains poorly addressed is uncovering the epigenetic regulatory mechanisms involved in the renewal and differentiation of dental pulp progenitors.
A highly conserved histone methyltransferase known as disruptor of telomeric silencing 1-like (Dot1L) plays a critical role in several cellular processes including cell cycle regulation, DNA damage repair, transcriptional elongation, cell differentiation and reprogramming (Sarno et al., 2020; Steger et al., 2008; Barry et al., 2009; Kim et al., 2014; Yang et al., 2019; FitzGerald et al., 2011; Wakeman et al., 2012). Dot1L itself is enriched at actively transcribed genes through its interaction with phosphorylated C-terminal domain of RNA polymerase II (Steger et al., 2008; Kim et al., 2012), and serves as the only enzyme that catalyzes the mono-, di- and tri methylation of histone 3 on lysine residue 79 (H3K79) (Feng et al., 2002). Enrichment of H3K79me2/3 within gene bodies is positively correlated with active transcription. Notably, a genome-wide analysis also revealed that H3K79me2 is deposited within gene regulatory sequences including enhancers and promoters (Wood et al., 2018; Ferrari et al., 2020). Mechanistically, Dot1L forms a complex with other factors such as AF10, AF17 and ENL to regulate transcription elongation in regions of active transcription. Recent studies suggest that Dot1L may also regulate transcriptional initiation by recruiting transcription factor IID (Wu et al., 2021). The interaction of Dot1L with mixed lineage leukemia (MLL) recruits RNA polymerase II to unmethylated CpG-rich promoters by forming a functional complex with the AF4/ENL/P-TEFb complex, MOZ and p300/CBP histone acetyl transferases (Miyamoto et al., 2020).
The essential role for Dot1L in mammalian development has been well-documented. Germline disruption of Dot1L and loss of H3K79me2 in mice results in mid-gestational lethality, which is attributed to defective extraembryonic vascular network formation, impaired hematopoietic development and cardiac hypertrophy (Duan et al., 2016; Feng et al., 2010). Multiple studies underscore the importance of meticulously regulated Dot1L function in establishing cell-type-specific transcriptional programs during differentiation. Several lines of evidence suggest that inhibition of Dot1L activity in progenitor populations biases the transcriptome from a stemness to a differentiation-mediating transcriptional program. For example, deficiency of Dot1L in the central nervous system reduces the number of neural progenitors, impairs neurogenesis and alters the distribution of neuronal subtypes (Franz et al., 2019; Bovio et al., 2019; Gray de Cristoforis et al., 2020). Yoo et al., determined that Dot1L function in blood endothelial cells or lymphatic progenitors ensures normal lymphatic development and function (Yoo et al., 2020). In the skeletal system, we previously demonstrated that early expression of Dot1L in limb mesenchymal progenitors provides regulatory control of endochondral bone growth and development through control of chondrocyte proliferation and differentiation (Sutter et al., 2021). Our research further implicated Dot1L in the regulation of key signaling pathways and associated genes involved in proliferation, as well as cell cycle checkpoint control in growth plate chondrocytes.
The epigenetic control of dental progenitors by Dot1L remains unexplored. Thus, in the present study, we performed single-cell RNA sequencing (scRNA-seq) to gain new insights into the mechanistic contributions of Dot1L in early postnatal mouse molar dental pulp. Our analysis generated transcriptomic signatures demarcating 15 distinct cell clusters within the molar dental pulp, including ameloblasts, vascular endothelial blood cells, pericytes and immune cells, as well as multiple subpopulations of MSCs at various stages of osteogenic differentiation. Notably, our analysis of the transcriptionally distinct cell populations that make up the mouse molar pulp revealed enrichment of Dot1L within cell clusters representing MSCs. We showed that Dot1L co-expresses with genes encoding the epigenetic factors Setd2, Sirt1, Suv39h1, Iws1, Pbrm1 and Bptf, which are associated with transcriptional activation. Interestingly, Dot1L co-expresses with genes encoding repressive epigenetic factors involved in DNA methylation and heterochromatin organization such as Dnmt1, Eed, Suv39h1 and Cbx3. We also noted that Dot1L co-expresses with downstream target genes associated with insulin growth factor (IGF) and WNT signaling and cell cycle progression. Taken together, our research provides novel insights into the role of Dot1L in the lineage commitment of MSCs in the mouse molar dental pulp.
Results
Single-cell RNA analysis of MSCs in the mouse molar dental pulp
We identified 15 distinct cell clusters in the dental pulp of mouse molars based on the differential expression of marker genes (summarized in Fig. 1A). Based on the expression of developmental genes required for skeletogenesis (Lmo4, Runx2, Sp7, members of the Lef1/Tcf family, Col1a1, Alpl, Bmpr1a, Fgfr3, Wnt5a, Tgfb2 and others) (Supplemental Fig. 1 and Supplemental Table 1), we identified 11 clusters of MSCs referred to as MSCs #1 to #10 and #13. Tooth development relies on reciprocal interactions between the ectoderm-derived dental epithelium and the underlying neural crest (NC)-originated mesenchyme (Hermans et al., 2021). In the mouse dental pulp, MSCs are derived from the cranial NC. A selective group of transcription factors known as NC master regulators, including Dlx, Msx, Sox, Id, Snai, Twist and others, control the NC specification program (Simoes-Costa and Bronner, 2015). We found that Dlx5, Twist1, Msx1, Msx2, Sox9 and members of the Id family are broadly expressed in all 11 subpopulations of MSCs (Supplemental Fig. 2).
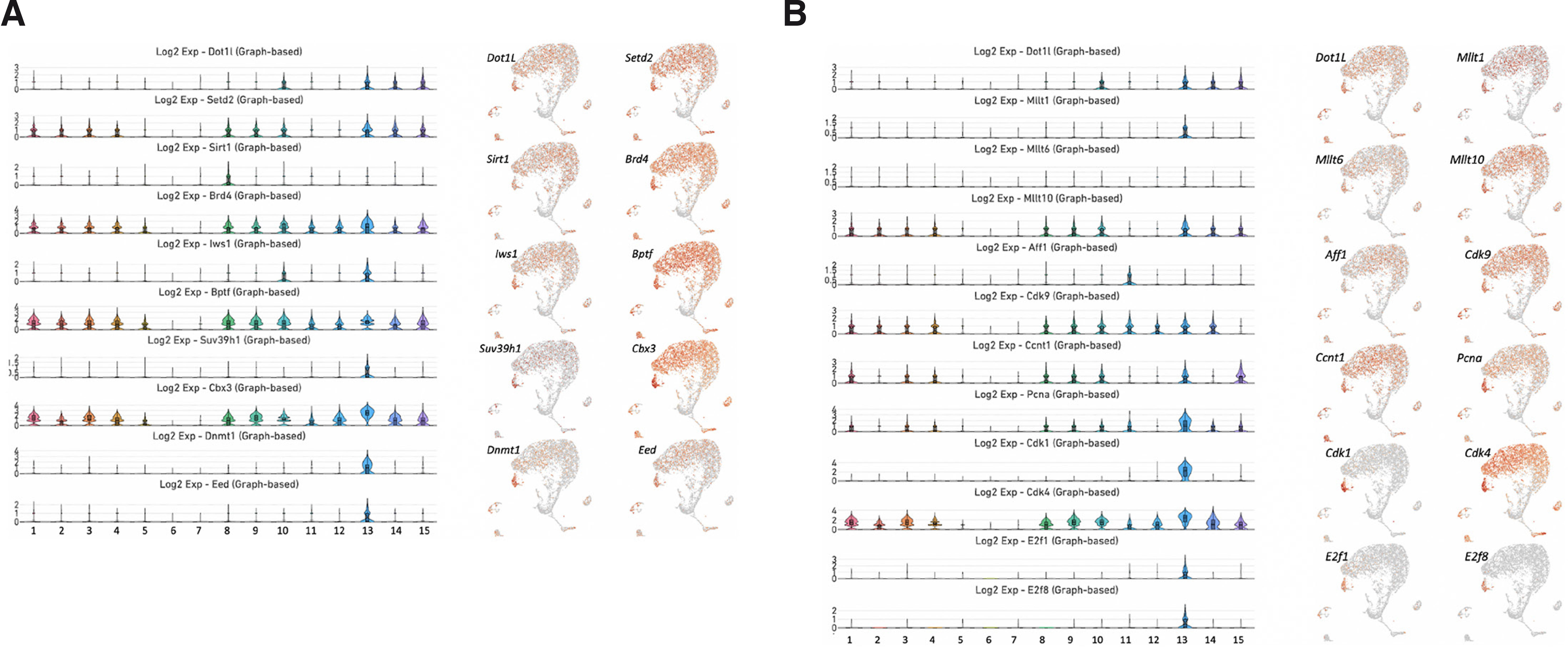
Fig. 1. scRNA-seq clustering analysis of the mouse molar dental pulp.
(A) The mouse molar dental pulp is composed of 15 distinct cell types that represent MSCs at different stages of osteogenic and odontogenic differentiation as well as pericytes, ameloblasts, immune cells and vascular endothelial cells. (B) STRING interaction network analysis revealed that Dot1L associates with SIRT1, SETD2, the MYC/p300 regulatory axis and β-catenin. SIRT1 physically interacts with SUV39H1, CBX3, DNMT1, NCOR1 and EED, key components of the repressive chromatin complexes. By contrast, SETD2 interacts with PBRM1, BPTF, NIPBL and IWS1, which are associated with transcriptional activation. The MYC/p300 regulatory axis controls the expression of Snai1, Zeb1 and Zeb2. Dot1L interacts with β-catenin, which regulates of the WNT signaling genes Tcf3, Tcf4 and Wnt5a.
Although previous reports have demonstrated that the dental pulp is composed of both fibroblasts and MSCs (Alvarez-Vasquez et al., 2022; Mollentze et al., 2021), we use MSCs as a common name to define both cell types. Recent studies have shown that MSCs and fibroblasts are identical with respect to their proliferation and differentiation capacities, gene expression profiles and immunomodulatory properties (Hematti, 2012; Denu et al., 2016; Soundararajan and Kannan, 2018), and it has been proposed that fibroblasts are in fact aged MSCs. Moreover, recent scRNA-seq studies have revealed significant similarities between cell subsets derived from MSCs and fibroblasts based on comprehensive analyses of integrated single-cell transcriptome data (Fan et al., 2022; Soliman et al., 2021). The authors suggested that MSCs represent a subclass of fibroblasts.
Differential expression of the osteogenic genes Lef1, Tcf7, Runx2, Sp7, Sp1, Snai1, Snai2, Twist2, Lmo4, Cebpb and Myc, which encode transcription factors, suggests that clusters 1 to 5 and 8 to 10 represent subpopulations of MSCs undergoing different phases of osteogenic differentiation. These clusters are also enriched in genes essential for skeletal development such as Alpl, Col1a1, Fgfr3, Wnt5a and Tgfb2 (Supplemental Fig. 1). Cluster 6 contains an undifferentiated population of stromal cells characterized by expression of Slc40a1, Cd55, Col3a1, Col5a2 and Col9a2 (Supplemental Table 1). In the adjacent cluster 7, we observed relatively modest expression of Alp, Col3a1, Fgfr1, Satb2, Id1, Id2 and Tgfb2 (Supplemental Fig. 1 and Supplemental Table 1). Within clusters 3 and 7, we identified a subpopulation of odontogenic cells enriched in Bglap, Dspp and Dmp1 (Supplemental Fig. 3).
There are some challenges to labeling stem cells in dental tissues using well-accepted markers such as Plp1 (Kaukua et al., 2014), Ng2/Cspg4 (Iwasaki et al., 2013), Gli1 (Zhao et al., 2014) and Sox2 (Sanz-Navarro et al., 2018). Although these markers are frequently used to trace stem cells, researchers have observed the expression of these genes in many tissues and cell types; more importantly, these markers are nearly ubiquitously expressed in skeletal lineages at different developmental stages (Ambrosi et al., 2019). Thus, based on the expression profiles of Plp1, Cspg4, Gli1 and Sox2, we cannot pinpoint a specific cluster as a population that represents stem cells, although the data hint that cluster 3 is likely to harbor a population of stem cells (Supplemental Fig. 4). Further research based on lineage tracing is required to define these cells.
Cluster 11 represents immune cells and is enriched in Fcer1g, Tyrobp, C1qb, Csf1r and Pf4 (Supplemental Fig. 5 and Supplemental Table 1). Cluster 12 defined pericytes, which express Rgs5, Ndufa4l2, Casq2, Acta2 and Myh1l (Supplemental Fig. 6 and Supplemental Table 1). We found that cluster 13 is enriched in a highly proliferative population of MSCs, as evidenced by an enrichment of genes involved in cell division or DNA replication (i.e., Nusap1, Spc25, Pimreg, Cenpf and Birc5) (Supplemental Fig. 7 and Supplemental Table 1). Cluster 14 represents ameloblasts based on the expressions of Krt5, Amelx, Fxyd3, Mmp20 and Enam (Supplemental Fig. 8 and Supplemental Table 1), whereas cluster 15 exhibits transcriptional profiles compatible with vascular endothelial cells (i.e., enrichment in Cdh5, Plvap, Ctla2a, Emcn and Cldn5) (Supplemental Fig. 9 and Supplemental Table 1).
Expression of Dot1L and epigenetic factors associated with Dot1L in the mouse molar dental pulp
The histone methyltransferase Dot1L regulates transcriptional initiation and elongation by recruiting RNA polymerase II to unmethylated CpG-rich promoters by forming a functional complex with different cofactors (Miyamoto et al., 2020). Protein interaction networks are critical for a system-level epigenetic understanding of gene regulatory processes. By analyzing the STRING interaction network (https://string-db.org), we discovered that Dot1L has a specific set of interacting partners (Fig. 1B). We identified SIRT1 and SETD2 as interacting partners of Dot1L. Our STRING analysis revealed that EED, a key component of the polycomb repressive complex 2 (PRC2), and the repressive proteins SUV39H1, CBX3 and DNMT1, which are known to be associated with heterochromatin and DNA methylation (Weirich et al., 2021; van Wijnen et al., 2021; Qin et al., 2011), can physically interact with SIRT1. In addition, Dot1L cooperates with the c-MYC-p300 complex to activate regulators of the epithelial-mesenchymal transition (Cho et al., 2015). The interaction of DOT1L with β-catenin and STAT1 ensures the proper control of the downstream targets of the WNT and JAK-STAT pathways (Monteagudo et al., 2017; Shah and Henriksen, 2011). Interestingly, although our uniform manifold approximation and projection (UMAP) analysis showed that Dot1L is broadly expressed in most clusters within the molar dental pulp, the violin plots clearly demonstrated an enrichment of Dot1L in clusters 10, 13, 14 and 15 (Fig. 2 and Supplemental Fig. 10). Next, we analyzed the expression of genes encoding critical protein partners of Dot1L. Setd2 was enriched in clusters 1, 2, 3, 4, 8, 9, 10, 13, 14 and 15. Sirt1 exhibited a more restricted expression with enrichment in cluster 8, while Suv39h1, Dnmt1 and Eed were mainly expressed in cluster 13 representing mitotic MSCs. Brd4, Bptf and Cbx were abundantly expressed in clusters 1, 2, 3, 4, 5, 8, 9, 10, 11, 12, 13, 14 and 15 (Fig. 2A and Supplemental Figs. 10 and 11). Dot1L participates in transcriptional elongation by forming a complex with AFF1, MLLT1, MLLT6 and MLLT10. We analyzed the expression pattern of genes encoding these factors. We detected expression of Mllt10 in clusters 1, 2, 3, 4, 8, 9, 10, 13, 14 and 15 (Fig. 2B and Supplemental Fig. 12). By contrast, Mllt1 was weakly expressed in many clusters although we observed some elevated expression in cluster 13. Mllt6 and Aff1 displayed relatively weak expression in all clusters.
Fig. 2. Expression of genes encoding epigenetic interactors, transcription elongation factors and downstream targets of Dot1L.
(A) Violin plots of the genes Dot1L, Setd2, Sirt1, Brd4, Iws1, Bptf, Suv39h1, Cbx3, Dnmt1 and Eed (left). UMAP visualization of genes encoding the components of the transcription activation complex, proteins involved in DNA methylation and components of the PRC2 (right). (B) Violin plots of the genes Dot1L, Mllt1, Mllt6, Mllt10, Aff1, Cctn1, Pcna, Cdk1, Cdk4, Cdk9, E2f1 and E2f8 (left). UMAP visualization of genes encoding proteins involved in transcription elongation and cell cycle progression (right).
Expression of downstream targets of Dot1L associated with cell cycle progression
Prior studies in erythroid progenitors, lung cells and chondrocytes reported that Dot1L loss of function induced decreased expression of genes involved in cell cycle regulation and proliferation (Kim et al., 2014; Sutter et al., 2021). Thus, we analyzed the expression profiles of cell cycle genes that are known downstream targets of Dot1L (Sutter et al., 2021). We found robust expression of Cdk4 and Cdk9 in clusters 1, 2, 3, 4, 8, 9, 10, 11, 12, 13 and 14 (Fig. 2B and Supplemental Figs. 12 and 13). In addition, Cdk4 was actively expressed in cluster 15. By contrast, expression of Cdk1, E2f1 and E2f8 was limited to cluster 13. Ccnt1 was enriched in clusters 1, 3, 4, 8, 9, 10, 13 and 15. Pcna exhibited a similar expression pattern with enrichment in clusters 1, 3, 4, 8, 9, 10, 11, 13, 14 and 15 (Fig. 2B and Supplemental Fig. 12).
Expression of cadherin and protocadherin genes, known downstream targets of Dot1L
Research indicates that depletion of Dot1L in Prrx1-expressing limb progenitors leads to dysregulation of cadherin and protocadherin genes (Sutter et al., 2021). Cadherin and protocadherin are at the crossroads of different signaling pathways (Pancho et al., 2020). Therefore, we performed a careful analysis of the expression profiles of these subfamilies in the mouse dental pulp. We found high levels of expression of Cdh2 and Cdh11 in clusters 1, 2, 3, 4, 5, 8, 9, 10, 12 and 13 (Fig. 3A). In addition, Cdk2 was expressed in cluster 14. By contrast, expression of Cdh3 and Cdh13 was limited to cluster 14 and cluster 15, respectively. Pcdh9 was enriched in clusters 1, 2, 3, 4, 5, 8, 9, 10 and 13. Pcdh10 was expressed in clusters 1, 4, 8, 9 and 10. Expression of Pcdh18 was limited to clusters 2 and 15. Pcdh18 was expressed in clusters 1, 3, 12 and 13, whereas Pcdh19 was restricted to cluster 12 (Fig. 3A).
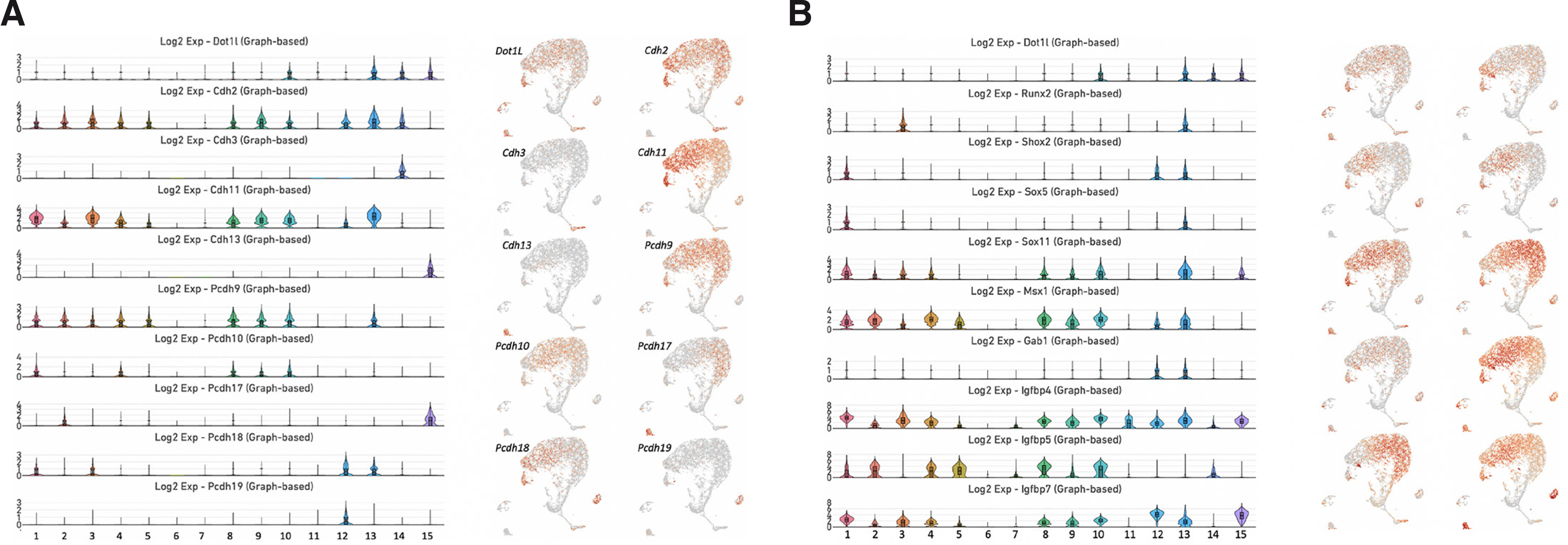
Fig. 3. Expression of genes encoding cadherins, protocadherins, osteogenic transcription factors and members of the IGF signaling pathway.
(A) Violin plots of the genes Cdh2, Cdh3, Cdh11, Cdh13, Pcdh9, Pcdh10, Pcdh17, Pcdh18 and Pcdh19 (left). UMAP visualization of genes encoding proteins involved in cell adhesion and cell-cell signaling (right). (B) Violin plots of the genes Runx2, Shox2, Sox5, Sox11, Msx1, Gab1, Igfbp4, Igfbp5 and Igfbp7 (left). UMAP visualization of these genes (right).
Expression of Dot1L transcriptional targets and downstream regulated pathways
We analyzed the expression profiles of genes encoding members of the IGF signaling pathway and some key transcription factors that were previously reported to be downstream targets of Dot1L in mouse limb chondrocytes (Sutter et al., 2021). The expression of Runx2 was limited to clusters 3 and 13 (Fig. 3B). Shox2 was expressed in clusters 1, 12 and 13, while Sox5 expression was restricted to clusters 1 and 13. By contrast, Sox11 displayed vigorous expression in clusters 1, 2, 3, 4, 8, 9, 10, 13, 14 and 15. Similarly, Msx1 was strongly expressed in clusters 1, 2, 3, 4, 5, 8, 9, 10, 13, 14 and 15. We identified high levels of Igfbp4 expression in clusters 1, 2, 3, 4, 5, 7, 8, 9, 10, 11, 12, 13, 14 and 15. Igfbp5 was expressed in clusters 1, 2, 4, 5, 7, 8, 9, 10 and 14, whereas Igfbp7 was more broadly expressed in clusters 1, 2, 3, 4, 5, 8, 9, 10, 12, 13 and 15 (Fig. 3B).
Ctnnb1 encoding β-catenin displayed a broad range of expression in the molar dental pulp (Fig. 4A). With the exception of clusters 6 and 7, Ctnnb1 was expressed in all other subpopulations of MSCs, as well as immune cells (cluster 11), pericytes (cluster 12), ameloblasts (cluster 14) and vascular endothelial cells (cluster 15). However, genes encoding transcription factors and ligands of the WNT signaling pathway exhibited a more restricted expression pattern. We noticed vigorous expression of Lef1, Tcf3, Tcf4, Tcf7, Wnt5a, Wnt6 and Wnt10a in cluster 13 (Fig. 4A). By contrast, expression of Tcf3, Tcf4 and Wnt5a was limited to cluster 10. The Tle1 gene, which encodes a member of the TLE/Groucho family of transcriptional corepressors, was mainly enriched in cluster 12 (pericytes).
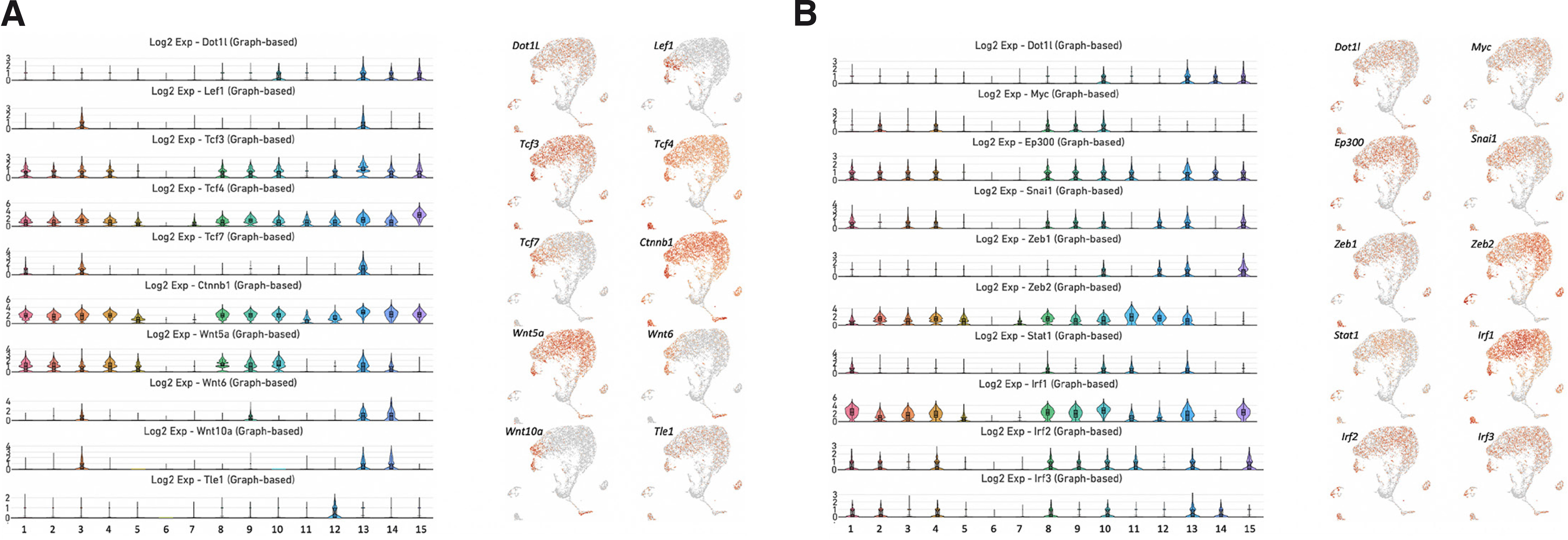
Fig. 4. Expression of genes encoding members of the WNT signaling pathway and the MYC/p300 regulatory axis.
(A) Violin plots of the genes Lef1, Tcf3, Tcf4, Tcf7, Ctnnb1, Wnt5A, Wnt6, Wnt10a and Tle1 (left). UMAP visualization of these genes (right). (B) Violin plots of the genes Myc, p300, Snai1, Zeb1, Zeb2, Stat1, Irf1, Irf2 and Irf3 (left). UMAP visualization of these genes (right).
Analysis of the expression patterns of the downstream targets of the Myc/p300 regulatory axis and JAK-STAT signaling pathway revealed enrichment of Ep300, Snai1, Zeb1 and Zeb2 in clusters 10 and 13, whereas c-Myc was enriched in cluster 10 only (Fig. 4B). Stat1 and transcription factors Irf1, Irf2 and Irf3 associated with the JAK-STAT pathway were actively expressed in clusters 10 and 13.
DNA demethylation, chromatin accessibility and RNA expression of Dot1L
We examined the genomic structure of Dot1L for the DNA demethylation mark 5-hydroxymethylcytocine (5hmC) and chromatin accessibility. 5hmC and open chromatin are hallmark features of genes that are actively transcribed. Using RNA-seq, assay for transposase-accessible chromatin using sequencing (ATAC-seq) and hydroxymethylated DNA immunoprecipitation sequencing (hMeDIP-seq) assays, we previously determined gene expression, accessible chromatin regions and genome-wide enrichment of 5hmC in the mouse dental pulp (Joshi et al., 2022a; Joshi et al., 2022b). By analyzing these datasets, we found that the genomic region across actively transcribed Dot1L gene exhibited open chromatin enriched in 5hmC (Fig. 5). Collectively, our results suggest that the Dot1L locus acquired an active chromatin state in the mouse dental pulp.
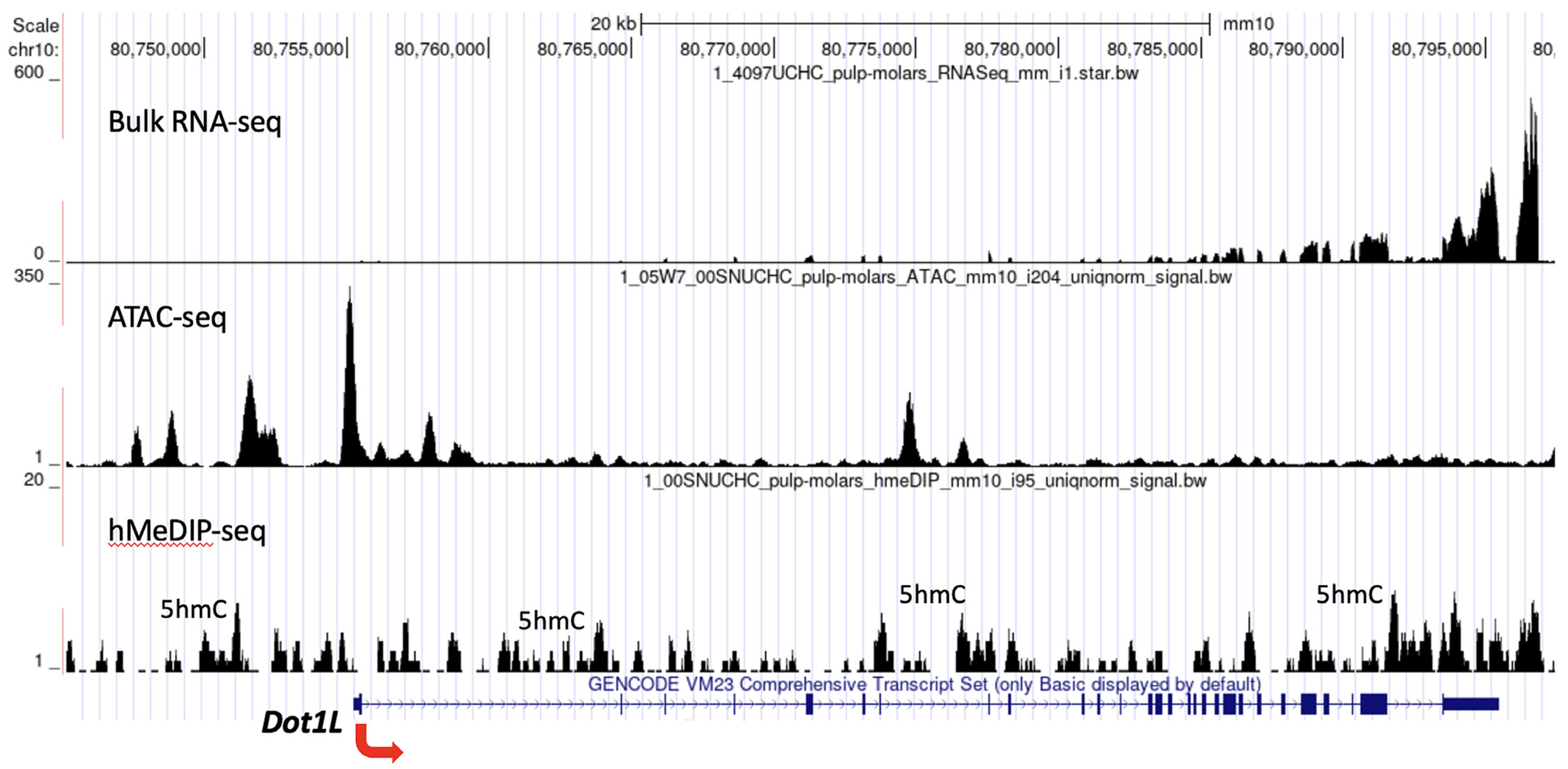
Fig. 5. Chromatin organization of Dot1L in the mouse molar dental pulp.
The genomic landscape of Dot1L in the mouse molar dental pulp. The hMeDIP-seq peaks are high in the promoter and gene body regions, indicating that 5hmC is enriched in the Dot1L locus. Strong ATAC-seq peaks are present in the transcription start site and promoter, which correlates with robust expression of Dot1L in the dental pulp.
Discussion
In the current study, we investigated the expression profile of Dot1L in the mouse molar dental pulp. Within 15 clusters of cells that represent pericytes, ameloblasts, vascular endothelial cells and MSCs, we found that Dot1L is significantly enriched in two subpopulations of MSCs at different stages of osteogenic differentiation (clusters 10 and 13), as well as in ameloblasts (cluster 14) and vascular endothelial cells (cluster 15).
We further investigated the expression patterns of components of the transcription initiation and elongation complexes associated with Dot1L in the mouse molar dental pulp. We identified a core set of genes that are common in these two clusters (Fig. 6). These genes encode SETD2, BRD4, IWS1, BPTF and MLLT10, which are key components of the transcription initiation and transcription elongation complexes. The mouse STRING database revealed that SIRT1 also interacts with EED, which is a subunit of the PRC2. In addition, SIRT1 forms a complex with factors involved in DNA methylation such as CBX3 and DNMT1. We analyzed the expression patterns of Eed, Cbx3 and Dnmt1 in the molar dental pulp and found that Cbx3 was expressed in both clusters, whereas Eed and Dnmt1 exhibited vigorous expression in cluster 13. Interestingly, cluster 13 represented mitotic MSCs that undergo cell division. The core set of transcription factors that were enriched in this subpopulation include Runx2, Shox2, E2F1, E2F8 and Sox5.
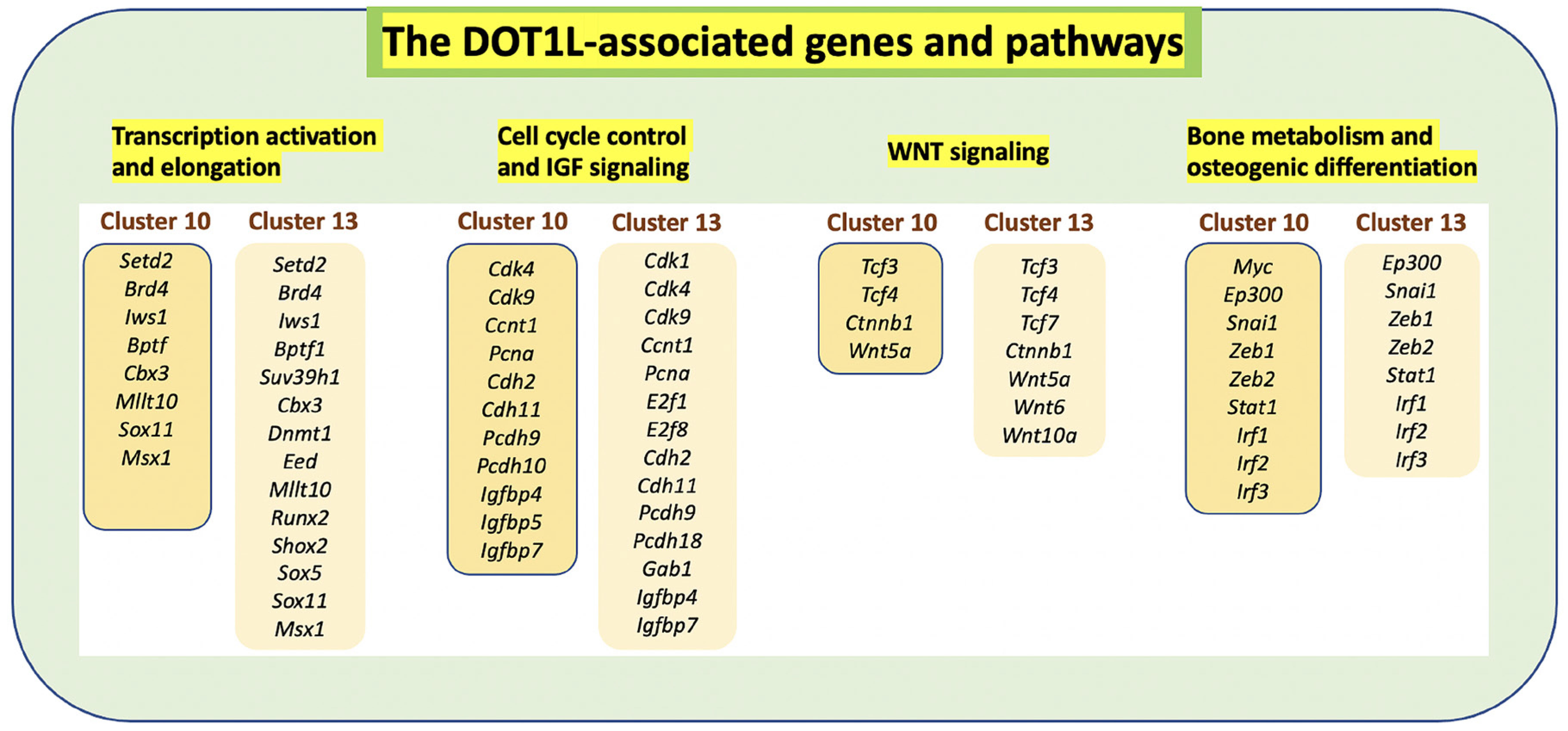
Fig. 6. Dot1L-associated genes and signaling pathways.
Dot1L is primarily expressed in clusters 10 and 13 representing MSCs at different stages of osteogenic differentiation. Dot1L associates with other components of transcription initiation and elongation to control the cell cycle and the expression of downstream targets of the WNT and IGF signaling pathways.
Surprisingly, Dot1L was not expressed uniformly across all identified MSCs (clusters 1–10 and 13). Some minor enrichment of Dot1L in the proliferating MSCs (cluster 13) within the mouse molar dental pulp is consistent with reports that Dot1L balances cell proliferation and differentiation by preventing premature cell cycle exit in progenitor populations (Nguyen and Zhang, 2011; Kim et al., 2014; Franz et al., 2019; Aslam et al., 2021). Although a major function of Dot1L is linked to cell proliferation (McLean et al., 2014), the impact of Dot1L depletion varies depending on the cell type or developmental stage (Kim et al., 2014). In previous studies, we showed that loss of Dot1L function in chondrocytes impaired transcriptional regulation of multiple genes implicated in cell cycle control and the IGF signaling pathway (Sutter et al., 2021). Our single-cell transcriptome analysis showed that Cdk4, Cdk9, Ccnt1, Pcna, Cdh2, Cdh11, Pcdh9, Igfbp4 and Igfbp7 were among the common genes enriched in clusters 10 and 13.
The expression of Dot1L in cluster 15 within the molar dental pulp is also congruent with published studies showing a mechanistic role for Dot1L in the development of the vasculature (Duan et al., 2016). Specifically, Duan et al., reported that Dot1L cooperates with the transcription factor ETS-1 to stimulate the expression of VEGFR2, thereby activating the ERK1/2 and AKT signaling pathways and promoting angiogenesis.
Dot1L is the only mammalian histone methyltransferase that catalyzes H3K79 methylation to mediate transcriptional activation (Vlaming and Leeuwen, 2016). Dot1L interacts with AF10, AF17 and ENL to regulate transcriptional elongation. Recent research showed that Dot1L is also involved in transcription initiation (Wu et al., 2021). Surprisingly, depletion of Dot1L and ENL reduced occupancy of the TATA-binding protein TBP and RNA polymerase II along target genes. Researchers have proposed that Dot1L may regulate transcription initiation by facilitating the recruitment of the transcription factor IID (Wu et al., 2021).
With the assistance of the histone H3K27 acetyltransferases CBP and p300, Dot1L is involved in sustaining cell-type-specific gene expression (Ebrahimi et al., 2019). H3K79 methylation mediated by Dot1L controls enhancer-promoter interactions and activation of specific marker genes in leukemia cells (Godfrey et al., 2021). However, during B-cell differentiation, Dot1L supports the repression of PRC2 targets associated with an antiproliferative plasma cell differentiation program (Aslam et al., 2021). Pharmacologic inhibition of Dot1L synergizes with the loss of SETD2 to induce growth arrest, DNA damage, differentiation and cell death (Skucha et al., 2018). Dot1L forms a complex with MLLT10/AF10 to bind TCF4/β-catenin in mouse small intestinal crypts (Mahmoudi et al., 2010). The recruitment of the MLLT10/AF10-Dot1L complex in a β-catenin-dependent manner results in H3K79 methylation and transcriptional elongation of WNT target genes. Surprisingly, Dot1L is also implicated in heterochromatin formation. Mouse embryonic stem cells (ESCs) deficient for Dot1l exhibit loss of H3K79 methylation accompanied by reduced heterochromatin marks H3K9me2 and H4K20me3, leading to proliferation defects, telomere elongation and aneuploidy (Jones et al., 2008).
While methylation of H3K79 is viewed as the sole molecular function of Dot1L activity, recent studies support methyltransferase-independent functions of Dot1L in ESCs, including transcriptional elongation and cellular differentiation (Cao et al., 2020). The global loss of Dot1L in mouse embryos firmly established the essential role of Dot1L in mammalian erythropoiesis (Feng et al., 2010), yet in vivo studies using a novel Dot1L methyltransferase mutant mouse model provided compelling evidence that Dot1L-controlled early embryonic erythropoiesis is independent of its intrinsic H3K79 methyltransferase activity (Malcom et al., 2022). These observations provide a strong rationale to thoroughly examine the functional contributions of Dot1L catalytic versus non-catalytic activities within various developmental processes and tissues, including the dental pulp. Some of the regulatory functions of Dot1L are likely mediated through its interactions with lineage-specific transcription factors or other chromatin remodeling complexes.
Our study revealed that Dot1l colocalizes with genes encoding protein interactors involved in epigenetic activation such as Setd2, Sirt1, Brd4, Isw1, Bptf and Suv39h1. In addition, we found that Dot1L is co-expressed with Eed2, Cbx3 and Dnmt1, which encode epigenetic factors associated with gene silencing and heterochromatin formation. Furthermore, we found that Dot1l is co-expressed with downstream targets of the IGF and WNT signaling pathways and cell cycle progression. Therefore, our findings suggest that Dot1L plays an important role in orchestrating lineage-specific gene expression during the differentiation of MSCs.
Collectively, our scRNA-seq analysis unmasked population heterogeneity of the mouse molar dental pulp. More importantly, our study provided invaluable insight into the single-cell gene expression pattern of Dot1L in osteogenic and odontogenic progenitors. Further investigation toward identifying the molecular pathways through which Dot1L histone methyltransferase orchestrates gene regulation will provide a more refined understanding of the fundamental mechanisms underlying the development of dental structures.
Materials and Methods
scRNA-seq and data analysis
We prepared primary pulp from the molars of 5- to 6-day-old mice according to previously described procedures (Balic et al., 2010). Viability of each single-cell suspension was assessed using a Countess II FL Automated Cell Counter (Thermo Fisher Scientific Inc., Waltham, MA). Suspensions of dissociated cells were loaded onto independent single channels of a Chromium Controller (10× Genomics, Pleasanton, CA) single-cell platform. Briefly, we loaded ~8,112 single cells for capture using a Chromium Single Cell 3′ Reagent kit, v2 Chemistry (10× Genomics). Following capture and lysis, complementary DNA was synthesized and amplified (14 cycles) as per the 10×Genomics protocol. The amplified cDNA was used to construct an Illumina sequencing library and was sequenced on a single lane of a HiSeq 4000 (Illumina, San Diego, CA).
Genome-wide ATAC-seq, hMeDIP-seq and RNA-seq
ATAC-seq, hMeDIP-seq and RNA-seq services were performed by Active Motif (Carlsbad, CA). RNA isolation was performed using the RNAeasy Mini/Midi kit (Qiagen, Germantown, MD). RNA-seq was performed using an Illumina NextSeq 500 to generate 42-nt paired-end sequences. For ATAC-seq and RNA-seq, the paired-end 42-bp sequencing reads generated by Illumina NextSeq 500 sequencing were mapped to the genome using the Burrows-Wheeler aligner (BWA) algorithm with default settings. For hMeDIP-seq, genomic DNA was isolated using the Monarch Genomic DNA Purification kit (New England Biolabs, Ipswich, MA) following the manufacturer’s instructions. DNA was sonicated to ~150–300 bp and Illumina adaptors were ligated to the DNA ends. To generate genome-wide maps of 5hmC, Active Motif performed hMeDIP-seq experiments using the antibody AM39791 to 5hmC. The input DNA was used as a control. Finally, immunoprecipitated DNA and input DNA that did not go through the immunoprecipitation step were processed into sequencing libraries and sequenced using the Illumina platform (NextSeq 500, 75-nt single-end).
Computational analysis
scRNA-seq
Analysis of scRNA-seq was completed using mouse molar dental pulp labeled as BD19002. The target was 6,000 cells, which produced an estimated 8,735 cells with 44,363 average reads per cell. Overall, the analysis utilized scanpy (Wolf et al., 2018), comprising several steps as detailed below. Quality control for the scRNA-seq FASTQ reads included:
-
(1) filtering FASTQ reads with more than 1 bp mismatched barcode (10X Genomics, 2018)
-
(2) filtering FASTQ reads with less than a 3 Q30 score
-
(3) filtering STAR alignments with less than a 255 MAPQ score (Dobin et al., 2013)
Samples were processed independently and then batch corrected via the BBKNN method (Polański et al., 2020). Single-cell clustering procedures do not perfectly partition cell types, but instead indicate similarity of expression patterns which can be useful for exploring specific gene profiles of genes such as Dot1L. The isolated cluster expression patterns can then be associated with cell mixtures by using cell-specific markers previously documented in the literature. Here, prior expression profiles derived from the 3,500 most variable dispersed genes were used to calculate the local neighborhood of the cells (Satija et al., 2015; Zheng et al., 2017). Next, a neighborhood graph was generated using the k-nearest neighbors (for k=30) algorithm, followed by dimensional reduction using the UMAP procedure (Becht et al., 2018). The neighborhood graph was then batch corrected with BBKNN (Polański et al., 2020). Single-cell clusters were assigned in the UMAP space according to the Leiden community detection method (Tragg et al., 2019). In addition to the above scanpy analysis, single-cell subclusters were generated visually when not automatically detected using the web-based Cellview software, which provides interactive viewing of the downstream data (Bolisetty et al., 2017). Figures generated for this work made use of screen captures of the Cellview interactions and supported the numerical results.
ATAC-seq
Genomic regions with high levels of ATAC-seq transposition/tagging events were determined using the MACS2 peak-calling algorithm (Zhang et al., 2008). Since both reads (tags) from paired-end sequencing represent transposition events, both were used for peak-calling but were treated as single, independent reads. To identify the density of the transposition events along the genome, the genome was divided into 32-bp bins and the number of fragments in each bin was determined. For this purpose, the reads were extended to 200 bp, which is close to the average length of the sequenced library inserts. In the default analysis, the tag number of all samples was reduced (by random sampling) to the number of tags present in the smallest sample. To compare peak metrics between two or more samples, overlapping intervals were grouped into “merged regions,” which were defined by the start coordinate of the most upstream interval and the end coordinate of the most downstream interval (to determine the union of overlapping intervals). In locations unique to a sample, the merged region was assigned to the interval. After defining the intervals and merged regions, the genomic locations and features were summarized and presented in Excel spreadsheets.
hMeDIP-seq
hMeDIP-seq reads were mapped to the mouse genome (mm10) using the BWA algorithm with default settings. Alignment information for each read was stored in the BAM format. For the analysis, only uniquely mapped reads without duplicates were used, and tag numbers were normalized to the lowest number among the samples (by downsampling), which was 19.7 million. Methylated regions (peak intervals) were identified using the MACS2 peak-calling algorithm (Zhang et al., 2008) with a default cutoff of p-value = 1e-7. The 5hmC tag distributions around the genes were determined and presented as average plots (average of values for all target regions).
Bulk RNA-seq
The 42 bp-long read-pair bulk RNA-seq fragments were mapped to the reference genome mm10 using the STAR aligner (Dobin et al., 2013). A fragment assignment step was carried out to count the number of fragments overlapping the genomic sequence. Only the read pairs that had both ends aligned at the same chromosome and the same DNA strand were considered for subsequent analyses. Feature counts (FPKM assignment to genes) were performed using the Subread package (Liao et al., 2014). Gene annotations were originally from the NCBI RefSeq database and then adapted by merging the overlapping exons from the same gene to form a set of disjoint exons for each gene. After obtaining the same table containing the fragments (or reads) of genes, differential analysis was performed using DESeq2 (Love et al., 2014).
Supplementary Material
Acknowledgements
This project was funded by the grant REP-401754-20144-20 from the University of Connecticut to DB. We thank all members of the MM, RG and DB laboratories. The data that support the findings of this study are available on request.
Declarations
Author contributions
DB: conceptualization. AV, BE and PR: methodology. DB, RG, TB, BE and PJ: data analysis. DB, PR and MM: reagents and materials. DB, RG, BE and TB: writing. DB, MM and DG: supervision. DB: funding acquisition. All authors gave final approval and agreed to be accountable for all aspects of the work.
References
Álvarez-Vásquez J. L., Castañeda-Alvarado C. P. (2022). Dental Pulp Fibroblast: A Star Cell. Journal of Endodontics 48: 1005-1019.
Ambrosi T. H., Longaker M. T., Chan C. K. F. (2019). A Revised Perspective of Skeletal Stem Cell Biology. Frontiers in Cell and Developmental Biology 7: 189.
Aslam M. A., Alemdehy M. F., Kwesi‐Maliepaard E. M., Muhaimin F. I., Caganova M., Pardieck I. N., Brand T., Welsem T., Rink I., Song J.Y., Wit E., Arens R., Jacobs H., Leeuwen F. (2021). Histone methyltransferase DOT1L controls state‐specific identity during B cell differentiation. EMBO reports 22: e51184.
Balic A., Aguila H. L., Caimano M. J., Francone V. P., Mina M. (2010). Characterization of stem and progenitor cells in the dental pulp of erupted and unerupted murine molars. Bone 46: 1639-1651.
Barry E. R., Krueger W., Jakuba C. M., Veilleux E., Ambrosi D. J., Nelson C. E., Rasmussen T. P. (2009). ES Cell Cycle Progression and Differentiation Require the Action of the Histone Methyltransferase Dot1L. Stem Cells 27: 1538-1547.
Bayarsaihan D., Enkhmandakh B., Vijaykumar A., Robson P., Mina M. (2022). Single-cell transcriptome analysis defines mesenchymal stromal cells in the mouse incisor dental pulp. Gene Expression Patterns 43: 119228.
Becht E., McInnes L., Healy J., Dutertre C.A., Kwok I. W. H., Ng L. G., Ginhoux F., Newell E. W. (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nature Biotechnology 37: 38-44.
Bolisetty Mohan T., Stitzel Michael L., Robson Paul, (2017). CellView: Interactive exploration of high dimensional single cell RNA-seq data. bioRxiv.org Preprint 123810.
Bovio P. P., Franz H., Heidrich S., Rauleac T., Kilpert F., Manke T., Vogel T. (2019). Differential Methylation of H3K79 Reveals DOT1L Target Genes and Function in the Cerebellum In Vivo. Molecular Neurobiology 56: 4273-4287.
Cao K., Ugarenko M., Ozark P. A., Wang J., Marshall S. A., Rendleman E. J., Liang K., Wang L., Zou L., Smith E. R., Yue F., Shilatifard A. (2020). DOT1L-controlled cell-fate determination and transcription elongation are independent of H3K79 methylation. Proceedings of the National Academy of Sciences 117: 27365-27373.
Cho M.H., Park J.H., Choi H.J., Park M.K., Won H.Y., Park Y.J., Lee C. H., Oh S.H., Song Y.S., Kim H. S., Oh Y.H., Lee J.Y., Kong G. (2015). DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nature Communications 6: 7821.
Denu R. A., Nemcek S., Bloom D. D., Goodrich A. D., Kim J., Mosher D. F., Hematti P. (2016). Fibroblasts and Mesenchymal Stromal/Stem Cells Are Phenotypically Indistinguishable. Acta Haematologica 136: 85-97.
Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29: 15-21.
Duan Y., Wu X., Zhao Q., Gao J., Huo D., Liu X., Ye Z., Dong X., Fu Z., Shang Y., Xuan C. (2016). DOT1L promotes angiogenesis through cooperative regulation of VEGFR2 with ETS-1. Oncotarget 7: 69674-69687.
Ebrahimi A., Sevinç K., Gürhan Sevinç G., Cribbs A. P., Philpott M., Uyulur F., Morova T., Dunford J. E., Göklemez S., Arı Sule, Oppermann U., Önder T. T. (2019). Bromodomain inhibition of the coactivators CBP/EP300 facilitate cellular reprogramming. Nature Chemical Biology 15: 519-528.
Fan C., Liao M., Xie L., Huang L., Lv S., Cai S., Su X., Wang Y., Wang H., Wang M., Liu Y., Wang Y., Guo H., Yang H., Liu Y., Wang T., Ma L. (2022). Single-Cell Transcriptome Integration Analysis Reveals the Correlation Between Mesenchymal Stromal Cells and Fibroblasts. Frontiers in Genetics 13: 798331.
Feng J., Mantesso A., De Bari C., Nishiyama A., Sharpe P. T. (2011). Dual origin of mesenchymal stem cells contributing to organ growth and repair. Proceedings of the National Academy of Sciences 108: 6503-6508.
Feng Q., Wang H., Ng H. H., Erdjument-Bromage H., Tempst P., Struhl K., Zhang Y. (2002). Methylation of H3-Lysine 79 Is Mediated by a New Family of HMTases without a SET Domain. Current Biology 12: 1052-1058.
Feng Y., Yang Y., Ortega M. M., Copeland J. N., Zhang M., Jacob J. B., Fields T. A., Vivian J. L., Fields P. E. (2010). Early mammalian erythropoiesis requires the Dot1L methyltransferase. Blood 116: 4483-4491.
Ferrari F., Arrigoni L., Franz H., Izzo A., Butenko L., Trompouki E., Vogel T., Manke T. (2020). DOT1L-mediated murine neuronal differentiation associates with H3K79me2 accumulation and preserves SOX2-enhancer accessibility. Nature Communications 11: 5200.
FitzGerald J., Moureau S., Drogaris P., O'Connell E., Abshiru N., Verreault A., Thibault P., Grenon M., Lowndes N. F. (2011). Regulation of the DNA Damage Response and Gene Expression by the Dot1L Histone Methyltransferase and the 53Bp1 Tumour Suppressor. PLoS ONE 6: e14714.
Franz H., Villarreal A., Heidrich S., Videm P., Kilpert F., Mestres I., Calegari F., Backofen R., Manke T., Vogel T. (2019). DOT1L promotes progenitor proliferation and primes neuronal layer identity in the developing cerebral cortex. Nucleic Acids Research 47: 168-183.
Godfrey L., Crump N. T., O’Byrne S., Lau I.J., Rice S., Harman J. R., Jackson T., Elliott N., Buck G., Connor C., Thorne R., Knapp D. J. H. F., Heidenreich O., Vyas P., Menendez P., Inglott S., Ancliff P., Geng H., Roberts I., Roy A., Milne T. A. (2021). H3K79me2/3 controls enhancer–promoter interactions and activation of the pan-cancer stem cell marker PROM1/CD133 in MLL-AF4 leukemia cells. Leukemia 35: 90-106.
Gray de Cristoforis A., Ferrari F., Clotman F., Vogel T. (2020). Differentiation and localization of interneurons in the developing spinal cord depends on DOT1L expression. Molecular Brain 13: 85.
Hematti P. (2012). Mesenchymal stromal cells and fibroblasts: a case of mistaken identity?. Cytotherapy 14: 516-521.
Hermans F., Hemeryck L., Lambrichts I., Bronckaers A., Vankelecom H. (2021). Intertwined Signaling Pathways Governing Tooth Development: A Give-and-Take Between Canonical Wnt and Shh. Frontiers in Cell and Developmental Biology 9: 758203.
Iwasaki K., Komaki M., Yokoyama N., Tanaka Y., Taki A., Kimura Y., Takeda M., Oda S., Izumi Y., Morita I. (2013). Periodontal Ligament Stem Cells Possess the Characteristics of Pericytes. Journal of Periodontology 84: 1425-1433.
Jones B., Su H., Bhat A., Lei H., Bajko J., Hevi S., Baltus G. A., Kadam S., Zhai H., Valdez R., Gonzalo S., Zhang Y., Li E., Chen T. (2008). The Histone H3K79 Methyltransferase Dot1L Is Essential for Mammalian Development and Heterochromatin Structure. PLoS Genetics 4: e1000190.
Joshi P., Vijaykumar A., Enkhmandakh B., Shin D.G., Mina M., Bayarsaihan D. (2022a). The chromatin accessibility landscape in the dental pulp of mouse molars and incisors. Acta Biochimica Polonica 69: 131-138.
Joshi P., Vijaykumar A., Enkhmandakh B., Mina M., Shin D.G., Bayarsaihan D. (2022b). Genome-wide distribution of 5hmC in the dental pulp of mouse molars and incisors. The Journal of Biochemistry 171: 123-129.
Kaukua N., Shahidi M. K., Konstantinidou C., Dyachuk V., Kaucka M., Furlan A., An Z., Wang L., Hultman I., Ährlund-Richter L., Blom H., Brismar H., Lopes N. A., Pachnis V., Suter U., Clevers H., Thesleff I., Sharpe P., Ernfors P., Fried K., Adameyko I. (2014). Glial origin of mesenchymal stem cells in a tooth model system. Nature 513: 551-554.
Kim W., Choi M., Kim J.E. (2014). The histone methyltransferase Dot1/DOT1L as a critical regulator of the cell cycle. Cell Cycle 13: 726-738.
Kim S.K., Jung I., Lee H., Kang K., Kim M., Jeong K., Kwon C. S., Han Y.M., Kim Y. S., Kim D., Lee D. (2012). Human Histone H3K79 Methyltransferase DOT1L Methyltransferase Binds Actively Transcribing RNA Polymerase II to Regulate Gene Expression. Journal of Biological Chemistry 287: 39698-39709.
Liao Y., Smyth G. K., Shi W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923-930.
Love M. I., Huber W., Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15: 550.
Mahmoudi T., Boj S. F., Hatzis P., Li V. S. W., Taouatas N., Vries R. G. J., Teunissen H., Begthel H., Korving J., Mohammed S., Heck A. J. R., Clevers H. (2010). The Leukemia-Associated Mllt10/Af10-Dot1l Are Tcf4/β-Catenin Coactivators Essential for Intestinal Homeostasis. PLoS Biology 8: e1000539.
Malcom C. A., Ratri A., Piasecka-Srader J., Borosha S., Chakravarthi V. P., Alvarez N. S., Vivian J. L., Fields T. A., Karim Rumi M.A., Fields P. E. (2022). Primitive Erythropoiesis in the Mouse is Independent of DOT1L Methyltransferase Activity. Frontiers in Cell and Developmental Biology 9: 813503.
Miyamoto R., Okuda H., Kanai A., Takahashi S., Kawamura T., Matsui H., Kitamura T., Kitabayashi I., Inaba T., Yokoyama A. (2020). Activation of CpG-Rich Promoters Mediated by MLL Drives MOZ-Rearranged Leukemia. Cell Reports 32: 108200.
Mollentze J., Durandt C., Pepper M. S. (2021). An In Vitro and In Vivo Comparison of Osteogenic Differentiation of Human Mesenchymal Stromal/Stem Cells. Stem Cells International 2021: 1-23.
Monteagudo S., Cornelis F. M. F., Aznar-Lopez C., Yibmantasiri P., Guns L.A., Carmeliet P., Cailotto F., Lories R. J. (2017). DOT1L safeguards cartilage homeostasis and protects against osteoarthritis. Nature Communications 8: 15889.
Nguyen A. T., Zhang Y. (2011). The diverse functions of Dot1 and H3K79 methylation. Genes & Development 25: 1345-1358.
Pancho A., Aerts T., Mitsogiannis M. D., Seuntjens E. (2020). Protocadherins at the Crossroad of Signaling Pathways. Frontiers in Molecular Neuroscience 13: 117.
Polański K., Young M. D., Miao Z., Meyer K. B., Teichmann S. A., Park J.E. (2020). BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics 36: 964-965.
Krivanek J., Soldatov R. A., Kastriti M. E., Chontorotzea T., Herdina A. N., Petersen J., Szarowska B., Landova M., Matejova V. K., Holla L. I., Kuchler U., Zdrilic I. V., Vijaykumar A., Balic A., Marangoni P., Klein O. D., Neves V. C. M., Yianni V., Sharpe P. T., Harkany T., Metscher B. D., Bajénoff M., Mina M., Fried K., Kharchenko P. V., Adameyko I. (2020). Dental cell type atlas reveals stem and differentiated cell types in mouse and human teeth. Nature Communications 11: 4816.
Qin W., Leonhardt H., Pichler G. (2011). Regulation of DNA methyltransferase 1 by interactions and modifications. Nucleus 2: 392-402.
Sanz-Navarro M., Seidel K., Sun Z., Bertonnier-Brouty L., Amendt B. A., Klein O. D., Michon F. (2018). Plasticity within the niche ensures the maintenance of a Sox2 + stem cell population in the mouse incisor . Development 145: dev155929.
Sarno F., Nebbioso A., Altucci L. (2020). DOT1L: a key target in normal chromatin remodelling and in mixed-lineage leukaemia treatment. Epigenetics 15: 439-453.
Satija R., Farrell J. A., Gennert D., Schier A. F., Regev A. (2015). Spatial reconstruction of single-cell gene expression data. Nature Biotechnology 33: 495-502.
Shah S., Henriksen M. A. (2011). A Novel Disrupter of Telomere Silencing 1-like (DOT1L) Interaction Is Required for Signal Transducer and Activator of Transcription 1 (STAT1)-activated Gene Expression. Journal of Biological Chemistry 286: 41195-41204.
Simões-Costa M., Bronner M. E. (2015). Establishing neural crest identity: a gene regulatory recipe. Development 142: 242-257.
Soliman H., Theret M., Scott W., Hill L., Underhill T. M., Hinz B., Rossi F. M.V. (2021). Multipotent stromal cells: One name, multiple identities. Cell Stem Cell 28: 1690-1707.
Soundararajan M., Kannan S. (2018). Fibroblasts and mesenchymal stem cells: Two sides of the same coin?. Journal of Cellular Physiology 233: 9099-9109.
Skucha A., Ebner J., Schmöllerl J., Roth M., Eder T., César-Razquin A., Stukalov A., Vittori S., Muhar M., Lu B., Aichinger M., Jude J., Müller A. C., Győrffy B., Vakoc C. R., Valent P., Bennett K. L., Zuber J., Superti-Furga G., Grebien F. (2018). MLL-fusion-driven leukemia requires SETD2 to safeguard genomic integrity. Nature Communications 9: 1983.
Steger D. J., Lefterova M. I., Ying L., Stonestrom A. J., Schupp M., Zhuo D., Vakoc A. L., Kim J.E., Chen J., Lazar M. A., Blobel G. A., Vakoc C. R. (2008). DOT1L/KMT4 Recruitment and H3K79 Methylation Are Ubiquitously Coupled with Gene Transcription in Mammalian Cells. Molecular and Cellular Biology 28: 2825-2839.
Sui B., Chen C., Kou X., Li B., Xuan K., Shi S., Jin Y. (2019). Pulp Stem Cell–Mediated Functional Pulp Regeneration. Journal of Dental Research 98: 27-35.
Sutter P. A., Karki S., Crawley I., Singh V., Bernt K. M., Rowe D. W., Crocker S. J., Bayarsaihan D., Guzzo R. M. (2021). Mesenchyme-specific loss of Dot1L histone methyltransferase leads to skeletal dysplasia phenotype in mice. Bone 142: 115677.
Traag V. A., Waltman L., van Eck N. J. (2019). From Louvain to Leiden: guaranteeing well-connected communities. Scientific Reports 9: 5233.
Tsutsui T. (2020). <p>Dental Pulp Stem Cells: Advances to Applications</p>. Stem Cells and Cloning: Advances and Applications Volume 13: 33-42.
van Wijnen A. J., Bagheri L., Badreldin A. A., Larson A. N., Dudakovic A., Thaler R., Paradise C. R., Wu Z. (2021). Biological functions of chromobox (CBX) proteins in stem cell self-renewal, lineage-commitment, cancer and development. Bone 143: 115659.
Vlaming H., van Leeuwen F. (2016). The upstreams and downstreams of H3K79 methylation by DOT1L. Chromosoma 125: 593-605.
Wakeman T. P., Wang Q., Feng J., Wang X.F. (2012). Bat3 facilitates H3K79 dimethylation by DOT1L and promotes DNA damage-induced 53BP1 foci at G1/G2 cell-cycle phases. The EMBO Journal 31: 2169-2181.
Weirich S., Khella M. S., Jeltsch A. (2021). Structure, Activity and Function of the Suv39h1 and Suv39h2 Protein Lysine Methyltransferases. Life 11: 703.
Wolf F. A., Angerer P., Theis F. J. (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biology 19: 15.
Wu A., Zhi J., Tian T., Cihan A., Cevher M. A., Liu Z., David Y., Muir T. W., Roeder R. G., Yu M. (2021). DOT1L complex regulates transcriptional initiation in human erythroleukemic cells. Proceedings of the National Academy of Sciences 118: e2106148118.
Yang L., Lei Q., Li L., Yang J., Dong Z., Cui H. (2019). Silencing or inhibition of H3K79 methyltransferase DOT1L induces cell cycle arrest by epigenetically modulating c-Myc expression in colorectal cancer. Clinical Epigenetics 11: 199.
Yoo H., Lee Y. J., Park C., Son D., Choi D. Y., Park J.H., Choi H.J., La H. W., Choi Y.J., Moon E.H., Saur D., Chung H. M., Song H., Do J. T., Jang H., Lee D. R., Park C., Lee O.H., Cho S.G., Hong S.H., Kong G., Kim J.H., Choi Y., Hong K. (2020). Epigenetic priming by Dot1l in lymphatic endothelial progenitors ensures normal lymphatic development and function. Cell Death & Disease 11: 14.
Zhang Y. D., Chen Z., Song Y. Q., Liu C., Chen Y. P. (2005). Making a tooth: growth factors, transcription factors, and stem cells. Cell Research 15: 301-316.
Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S. (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biology 9: R137.
Zhao H., Feng J., Seidel K., Shi S., Klein O., Sharpe P., Chai Y. (2014). Secretion of Shh by a Neurovascular Bundle Niche Supports Mesenchymal Stem Cell Homeostasis in the Adult Mouse Incisor. Cell Stem Cell 14: 160-173.
Zhao L., Zhang P., Galbo P. M., Zhou X., Aryal S., Qiu S., Zhang H., Zhou Y., Li C., Zheng D., Bhatia R., Lu R. (2021). Transcription factor MEF2D is required for the maintenance of MLL-rearranged acute myeloid leukemia. Blood Advances 5: 4727-4740.
Zheng G. X. Y., Terry J. M., Belgrader P., Ryvkin P., Bent Z. W., Wilson R., Ziraldo S. B., Wheeler T. D., McDermott G. P., Zhu J., Gregory M. T., Shuga J., Montesclaros L., Underwood J. G., Masquelier D. A., Nishimura S. Y., Schnall-Levin M., Wyatt P. W., Hindson C. M., Bharadwaj R., Wong A., Ness K. D., Beppu L. W., Deeg H. J., McFarland C., Loeb K. R., Valente W. J., Ericson N. G., Stevens E. A., Radich J. P., Mikkelsen T. S., Hindson B. J., Bielas J. H. (2017). Massively parallel digital transcriptional profiling of single cells. Nature Communications 8: 14049.