Int. J. Dev. Biol. 66: 359 - 372 (2022)
Regeneration and proliferation of cardiomyocytes and its microRNA regulatory mechanisms
Review | Published: 22 December 2022
Abstract
Myocardial regeneration is identified as a concept at histological level. The core content is to increase the number of cardiomyocytes (CMs), so as to maintain the steady state of CMs under pathological or physiological conditions and ensure the normal cardiac function. In this review, we discussed the relevant factors involved in the regeneration of CMs, generalized in mice, large mammals and human. During different development stages of mammalian hearts, CMs showed several controlling and growth modes on the physiological or pathological state: mitosis, hypertrophy, nuclear polyploidy and multinucleation, amitosis and etc. We also discussed the mechanisms of specific microRNAs implicated in the cardiac development, as well as disease-induced apoptosis in CMs and the process of re-entering cell cycle after injury. It is hoped that this review will contribute to a deeper understanding of therapeutic approaches for myocardial regeneration after injury.
Keywords
cardiomyocytes, regeneration, proliferation, myocardial infarction, miRNA
Introduction
The heart comprises parenchyma (cardiac myocytes) and stroma (connective tissue). Connective tissue was considered inert, but some evidence revealed that stroma and its cellular constituents were dynamic and metabolically active to turn over the fibrillar collagen by regulating the composition of self-peptide hormones (Weber et al., 1995). The cardiomyocytes (CMs) are the dominant cell components of myocardium (Sylva et al., 2014). Notably, one gram loss of CMs per year in physiological condition was associated with aging (Olivetti et al., 1991), concomitant with the formation of irreversible fibrotic scar tissue lacking contractile properties (Gamba et al., 2014). Following multiple factors, the decrease in CMs occurred in several pathological processes, including myocardial infarction (MI). In the human left ventricular myocardium, there exists about 2-4 billion CMs, of which 25% slashed in a few hours after MI takes place (Murry et al., 2006). In hearts subjected to pressure overload of the left ventricle induced by hypertension or other cardiovascular diseases, the decreased number of CMs and excessive fibroblast accumulation were responsible for fibrosis of the myocardium, a major clinical issue (Whelan et al., 2010). A critical cause of cardiovascular diseases mortality is the cardiomyocyte’s inability to renew after cardiac injury. The fundamental strategies for cardiac regeneration and repair have kept investigators pursuing them for years. The relevant therapeutic approaches have focused on replacing dead or dying CMs, for instance, by autologous cell transplantation of the orientational differentiation to CMs from different stem cells (Terzic and Behfar, 2016), in parallel with unclear benefit, mode of action, and potential side effects. An alternative target is the stimulation of substantial regeneration after injury through cardiomyocyte proliferation (Mahmoud et al., 2013). microRNAs (miRNAs) are a batch of single-stranded noncoding RNAs which play an essential role in regulating the transcription of gene expression level (Ameres and Zamore, 2013). A growing body of evidence indicated that endogenous capacity of regeneration can be achieved by exogenously delivering miRNAs after MI (Eulalio et al., 2012; Migliore et al., 2008). miRNAs light on exploring interventional approaches for preventing heart failure or treating heart failure therapeutically in clinical trials, thus enhancing the heart’s function and prolonging the human life span. As a promising strategy to supplement functional myocytes for cardiac repair, there is a pressing need to understand the proliferation modes and relevant miRNA regulatory mechanisms of heart regeneration. Herein, we systematically reviewed the proliferation modes of CMs and well-established and emerging roles of miRNAs in cardiac development, aiming to provide a potential therapeutic target by manipulating the miRNA network for cardiac regeneration.
An overview of miRNA
miRNAs, a batch of single-stranded noncoding RNAs about 18–22nt long, play an essential role in regulatinggene expression at the post-transcriptional level (Ameres and Zamore, 2013). miRNA biogenesis often begins with RNA polymerase II transcription of primary miRNA (pri-miRNA). Unlike protein-coding genes, miRNA transcription start sites can be more than several kilobases upstream of mature miRNAs (Wang et al., 2021). Following the transcription, the long pri-miRNAs are precisely cropped around a hairpin-shaped region by a microprocessor consisting of the enzymes Drosha ribonuclease III and its cofactor protein DGCR8 to generate precursor miRNAs (pre-miRNA) (Wang et al., 2021). Pre-miRNAs are then processed by the enzyme Dicer 1 ribonuclease III, recruiting Argonaute proteins (AGO2) and transactivation response element RNA-binding protein, mediating the assembly of the RNA-induced silencing complex (RISC) where the mature miRNA is then generated following the removed passenger strand of the miRNA duplex (Chekulaeva and Filipowicz, 2009). In combination with the RISC, the mature miRNA bound the 3' untranslated regions of mRNA by forming complementary base pairing and blocked the translation of mRNA or degraded the mRNA to silence its targeted genes by recruiting AGO2 to mRNA targets (Sheu-Gruttadauria and MacRae, 2018). Occasionally, miRNAs constructed a complicated network of interactions by binding to the 5′-UTR, promoter, or the open reading frame regions (Lagos-Quintana et al., 2001). Intriguingly, miRNAs target more than 60 % of protein-coding regions in the human genome, suggesting the widespread effects of miRNAs on transcriptome and proteome diversity as well as function in a context-dependent manner (Lagos-Quintana et al., 2001; Li et al., 2022).
It is of note that miRNA play a pivotal role in modulating the initiation and progression of cardiovascular diseases by regulating different molecular mechanisms. For instance, the up-regulation of miR-29 could reduce lesion size and necrotic zones via targeting Col1A and Col3A (Ulrich et al., 2016). In addition, long noncoding RNAs (lncRNAs), serving as miRNA sponges and the upstream negative regulator of the miRNA/mRNA axis, regulate the biological activities of cardiomyocytes and cardiac fibroblasts. The hypertrophy of CMs may lead to impairment of cardiac function. The lncRNA AK048451, named cardiac hypertrophy-related factor, was identified as an endogenous sponge of miR-489 and promoted cardiac hypertrophic effects by regulating the miR-489 activity and the expression of its target (Wang et al., 2014a).
Additionally, a myocardial infarction-associated transcript was confirmed as a binding site for miR-150, modulating proliferation, migration, and apoptosis (Shen et al., 2016). Mitochondrial fission and fusion are also related to cardiomyocyte apoptosis. miR-539 could directly interact with lncRNA AK017121, thus reducing cardiomyocyte apoptosis mediated by mitochondrial fission and providing a novel approach for the treating of ischaemic heart disease (Wang et al., 2014b). The miRNAs also participate in the autophagy of cardiomyocytes, an evolutionarily conserved process in response to stress stimuli. miR-188-3p, modulated by lncRNA AK079427, targeted ATG7 to reduce autophagic vesicles formation and cell death in cardiomyocytes and thus viewed as a therapeutic target to preserve cardiac function (Wang et al., 2015b). Cardiomyocyte necrosis mainly contributes to heart failure (HF). The inhibition of miR-103/107 and its target Fas-associated protein with the death domain in response to H2O2 treatment by lncRNA H19 prevented cardiomyocyte necrosis (Wang et al., 2015a). miRNAs also involve in the activation of cardiac fibroblast. The overexpression of GAS5 prevented the growth of cardiac fibroblasts. It inhibited the expressions of Col1A1 and α-SMA at both the mRNA and protein levels via negatively regulating miR-21 and its target, phosphatase, and tensin homolog (Tao et al., 2017). Expression of miRNAs can be achieved by gene therapy using adeno-associated vectors, which transduce cardiomyocytes with high efficiency (Zacchigna et al., 2014). The clinical application of miRNA required more extensive experimentation in large mammals and rigorous safety assessment.
Regeneration of mammalian myocardium
Adult zebrafish retain a robust capacity for cardiac regeneration throughout life: the pre-existing CMs in the heart of zebrafish dedifferentiated and entered the cell cycle after injury (Genge et al., 2016; Tahara et al., 2016). The type of cardiac regeneration in zebrafish left no scar even after up to 20% resection of the ventricle (Poss et al., 2002). Did this type of regeneration occur in the mammalian heart?
Mouse
To date, several studies have confirmed the regeneration capacity of CMs after injury in mice. An impressive regenerative capacity of fetal heart was shown to compensate for an effective loss of 50% of cardiac tissue (Drenckhahn et al., 2008). Consistently, the hearts of 1-day-old neonatal mice were shown to regenerate after partial surgical resection characterized by minimal hypertrophy or fibrosis. However, this capacity was switched off early by seven days of age. Of note, regenerated tissue originated from pre-existing CMs was detected by genetic fate mapping (Porrello et al., 2011b). In 2012, Haubner et.al presented for the first time a mammalian model of complete cardiac regeneration following a severe ischemic cardiac injury (Haubner et al., 2012). Subsequently, Polizzotti et al., presented an alternative model for the study of cardiac regeneration in neonatal mice in which cryoinjury was used to induce heart injury (Polizzotti et al., 2016). Of note, severe systemic hypoxemia induced a robust regenerative response with decreased myocardial fibrosis and improvement of left ventricular systolic function in adult mice (Nakada et al., 2017), lighting on the potential therapeutic role of hypoxia in regenerative medicine.
So up to now, several studies have revealed the mechanisms required for regenerative program’s activation using the mouse neonatal heart regeneration model. The downregulation of low-density lipoprotein receptor-related protein 6 increased cardiomyocyte cell cycle activity in neonatal, juvenile, and adult mice (Wu et al., 2021). Nrf1, a stress-responsive transcription factor, was found to promote heart regeneration and repair by regulating proteostasis and redox balance using the mouse neonatal heart regeneration model (Cui et al., 2021). Remarkably, the loss of regeneration capacity within the first week of life coincided with a metabolic shift from glycolysis to oxidative phosphorylation, thus inducing significantly increased reactive oxygen species production and DNA damage. The utilization of fatty acids exerted adverse effects on cardiomyocyte proliferation in this model, which is emerging as a viable target for cardiac regenerative therapies at the metabolic level (Cardoso et al., 2020). Furthermore, succinate dehydrogenase inhibition in neonatal mice exerted positive effects on adult cardiomyocyte proliferation, revascularization, and heart regeneration via metabolic reprogramming (Bae et al., 2021).
Large mammals
So far, the large mammal model, including pigs, dogs, and sheep, is an urgent pre-clinical need to advance our knowledge of cardiovascular disease and to explore new approaches to repair the damaged heart (Camacho et al., 2016; Velayutham et al., 2019). Exosomes secreted by cardio-sphere-derived cells were shown to reduce scarring, attenuate adverse remodeling, and improve function in acute and chronic MI in the porcine heart (Gallet et al., 2017). Neonatal porcine hearts were capable of regeneration after MI, likely driven by cardiac myocyte division, but this potential was immediately lost when CMs exited the cell cycle shortly after birth (Ye et al., 2018; Zhu et al., 2018). This regenerative capacity was abrogated by corticosteroid in neonatal pig hearts (Tao et al., 2020). Subsequently, the delivery of human miRNA-199a in infarcted porcine hearts was shown to stimulate cardiac repair with marked improvements in global and regional contractility, increased muscle mass, and reduced scar size (Gabisonia et al., 2019). These results show that achieving cardiac repair through the stimulation of endogenous cardiomyocyte proliferation is attainable in large mammals. Like adult mice, agrin was capable of reducing ischemia-reperfusion injury and improving heart function in a pre-clinical porcine model (Baehr et al., 2020). The relevant application has focused on the transplantation of human CMs into the pig model, for instance, by transplanting immature human embryonic stem cell-derived CMs in the infarcted porcine heart (Romagnuolo et al., 2019), providing new approaches to testify the mechanistic basis of electrical instability following exogenous CM transplantation in humans.
Human
Over the past three decades, there was a relatively small number of case reports supporting that patients suffering from MI in the neonatal period, without structural heart diseases, suggests that humans owned the capacity for heart regeneration after birth (Boulton et al., 1991; Cesna et al., 2013; Deutsch et al., 2014; Farooqi et al., 2012; Murugan et al., 2002; Peeters et al., 1993). Saker and colleagues reported a complete myocardial function recovery after massive cardiogenic shock in a neonate within the first day of delivery, without affecting the neurodevelopment of the neonate (Saker et al., 1997). Subsequently, a newborn child having a severe myocardial infarction due to coronary artery occlusion was observed to have functional cardiac recovery, which translated into long-term normal heart function (Haubner et al., 2016). Considering all the evidence, newborn humans, similar to neonatal rodents, might have the intrinsic capacity to repair myocardial damage and recover cardiac function completely. In addition, four adult patients suffering anomalous left coronary artery from the pulmonary artery had no evidence of myocardial scarring after corrective surgery (Fratz et al., 2011), suggesting the possibility of heart regeneration in adults. In addition, endothelial cells in the heart were shown to have turnover rates and, early childhood possessed the highest turnover rate of CMs (Bergmann et al., 2009; Bergmann et al., 2015). The potential regeneration capacity in the adult human heart suggests that it may be rational to work toward the development of therapeutic strategies focused on stimulating this process in cardiac pathologies.
Taken together, fundamental insights gained from cardiac repair in neonatal mice and large mammals generate fresh insights into future cardiac regeneration in humans. Although the observations reported so far hint at the possibilities of cardiac repair in human infants, therestill needs to be more cases to map the time window for cardiac functional recovery. Additionally, functional cardiac recovery was observed in infants following a massive infarction, and there remains a lack of evidence on whether it can also occur after permanent coronary vessel occlusion, as in the rodent MI models, or rely on reperfusion. Considering the nature of the case report, the intrinsic capacity to regenerate cannot directly be proved at a cellular level.
Given all that has been mentioned above, it can be assumed that mammals might have the intrinsic capacity to regenerate their hearts through proliferating and dividing despite doing so at a slow rate. Reliable determination of cardiomyocyte proliferation is challenging. The various models used each have specific shortcomings that undermine definitive conclusions (Table 1). Additionally, the revelation of the proliferation modes of CMs and the related regulatory mechanisms with significant therapeutic value for MI were also discussed below.
Table 1
Mammalian models used in the research of cardiomyocytes
| Model | Anatomical Characteristics of Heart | Advantages | Disadvantages |
|---|---|---|---|
| Neonatal Mice | Four chambers and a closed circulation with capillaries (Bettex et al., 2014). | Mice have similarities in evolution with humans making it a more clinical model (Porrello et al., 2013). |
1. Mice have a period limitation within the first week after birth (Porrello et al., 2011b). 2. Mice own unclear ‘regenerative window’ (Uygur and Lee, 2016). |
| Neonatal Pigs |
A classic 'Valentine heart' shape (Crick et al., 1998); Two pulmonary veins into the left atrium (Crick et al., 1998); Aortic-mitral fiber with less continuity than humans (Crick et al., 1998). |
The application of large mammals is closer to the clinic (Ye et al., 2018). |
1. The use of large mammals makes high expenditure (Ye et al., 2018); 2. Lack of tools to manipulate genetically (Ye et al., 2018). 3. CMs grow primarily by multinucleation and longitudinal hypertrophy in an older pig, distinct from mice and humans(Velayutham et al., 2020). |
| Neonate |
Low pulmonary circulation pressure (Tan and Lewandowski, 2020); High systemic circulation resistance (Tan and Lewandowski, 2020); Foramen ovale and Ductus arteriosus closed (Saker et al., 1997). |
Possess the highest value for transfer to clinical practice (Levy et al., 2018). |
1. Utilizing neonates as research models have ethical issues (Mackintosh and Armstrong, 2020). 2. Regeneration critically depends on the types of injuries (Sen and Sadek, 2015). 3. There exist various regulators of postnatal cell cycle arrest (Nakada et al., 2015). |
Proliferation modes of mammalian cardiomyocytes
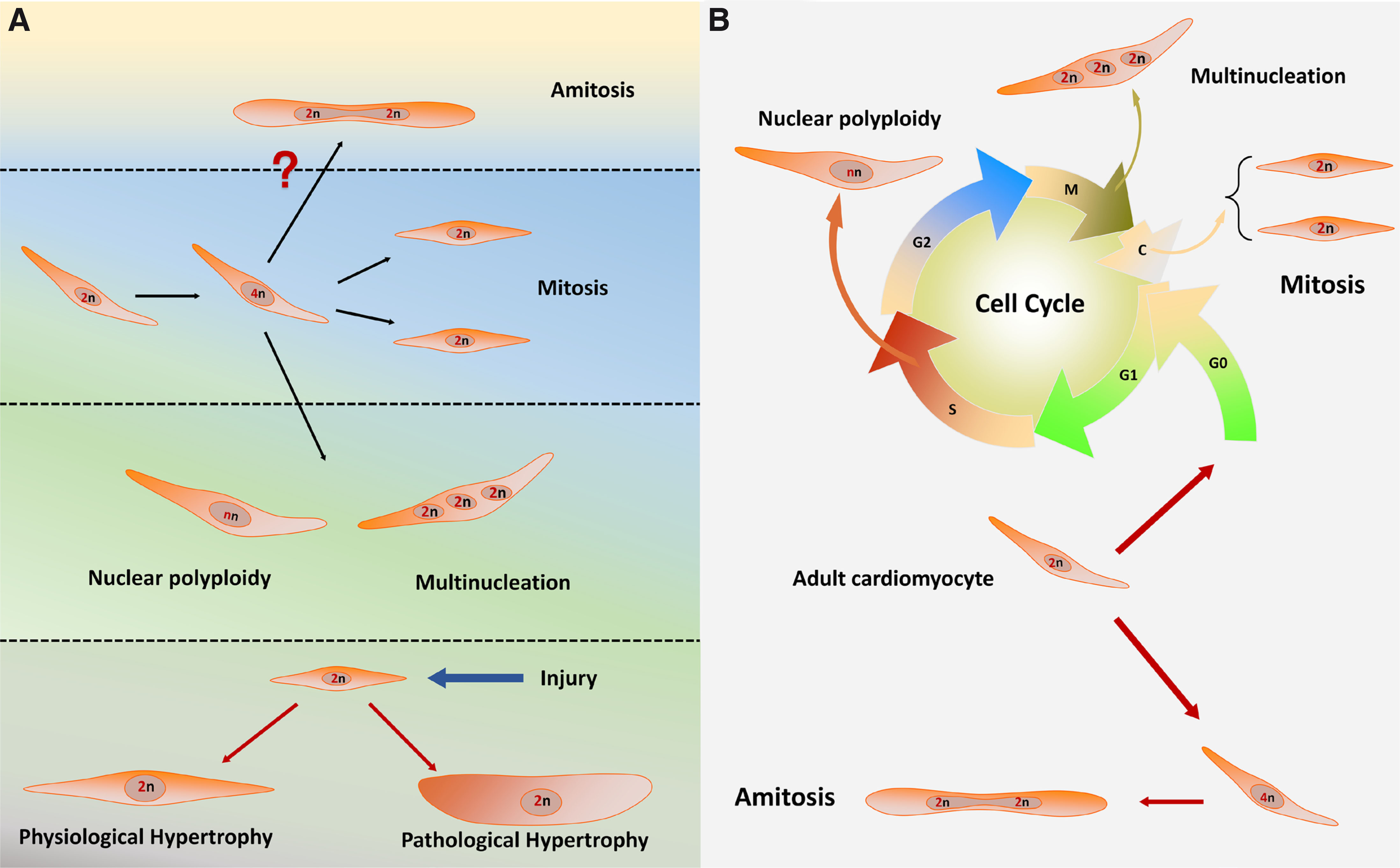
Fig. 1. Different growth and proliferation modes of cardiomyocytes
(A) The four growth modes of cardiomyocytes and their relevant developing stages are shown. (B) The growth modes of cardiomyocytes are divided into the mitosis and amitosis processes. The cell cycle stages when mitotic growth modes happen are shown, and the amitotic process is also exhibited. The “n” in the nucleus of cardiomyocytes means the number of homologous chromosomes. “?” means the indefinite existence of amitosis.
Mitosis
CMs undergo nuclear division at high speed during embryogenesis; however, human heart regeneration becomes limited to very slow cardiomyocyte replacement. Mitosis occupies only ~2% of the cell cycle, making it hard to quantify meaningfully. It is reported that mitotic cells existed in the normal cardiomyocytes in prenatal and postnatal ratsbut, in parallel, decreased along the developing stages (Guo et al., 2011). Similarly, in a rat model of complete cardiac regeneration following a severe ischemic cardiac injury, a transcriptional program of significant changes in genes mediating mitosis and cell division between days 1, 3, and 10 postnatal was observed (Haubner et al., 2012). In a study investigating healthy human hearts aged 0-59 years, cardiomyocyte mitosis was detectable throughout life; in contrast, cardiomyocyte cytokinesis was not evident after 20 years, suggesting cardiomyocyte proliferation contributed to developmental heart growth in young humans (Mollova et al., 2013). The overexpression of cyclin-dependent kinase 1 (CDK1), CDK4, cyclin B1, and cyclin D1 induced high CM mitotic activity in post-mitotic mouse, rat, and human cardiomyocytes, which was assessed by co-localization of histone H3 phosphorylation (marking cells that may enter mitosis) staining with cardiac Troponin-T staining (Mohamed et al., 2018). In addition, following activation of E2F1 and E2F3, CMs were induced into DNA synthesis as well as increased apoptosis without actual mitosis and cell division (Ebelt et al., 2005), which indicated that targeting a single factor, either positively or negatively, was not sufficient to achieve complete cell replication.
Various methods were used for the detection of mitotic CMs. The most solid evidence to demonstrate incontrovertibly for the first time that CMs are generated after birth was carbon-14 (14C) dating of cTropTþ nuclei. The elevated atmospheric 14C levels were generated by nuclear bomb tests during the Cold War and taken up by plants (Levin I et al., 2010). After eating these plants, the 14C was integrated into the synthesized genomic DNA, thus establishing the age of CMs in humans (Spalding et al., 2005a). By correlating levels of 14C in cardiomyocyte nuclei with atmospheric 14C levels in different years, Bergmann et al., reported that CMs renewed, with a gradual decrease from 1% turning over annually at the age of 25 to 0.45% at the age of 75. Additionally, less than half of CMs were exchanged in the life cycle (Bergmann et al., 2009). Notably, the resolution of the 14C dating is about 1-2 years, which makes it unsuitable for species with a short lifespan (Spalding et al., 2005b). Despite this effective method, which indirectly correlated nuclear division with cell division, the analysis of cell division is complicated by cardiomyocyte binucleation shortly after birth and the difficulties in recognizing cardiomyocyte nuclei in conventional histological sections. While the low and discrete rate of cardiomyocyte generation in mice was calculated by combining two different pulse-chase approaches (Senyo et al., 2013), a systematic understanding of the extent of division in the postnatal mammalian hearts was still lacking. Using the mosaic analysis with double markers (MADM) mouse model, Ali et al., provided crucial proof for the differentiated α-myosin heavy chain-expressing cardiomyocyte as the original cell of postnatal cardiomyogenesis (Ali et al., 2014), suggesting postnatal cardiomyogenesis in a cellular level.
Hypertrophy
Hypertrophic growth relates to cardiac pathologies with expansion in size and increased amounts of connected sarcomeres. This process maintains the structural integrity and contractile force of the heart wall responding to the changing mechanical workload (Baidyuk et al., 2019). Consistently, cardiac hypertrophy is concomitant with changes in gene expression, thus inducing changes in metabolism, contractility, and cardiomyocyte survival. There are two types of hypertrophy: physiological and pathological. Both types are initially emerging as an adaptive response to stress. Physiology hypertrophy exhibits a mild increase in cardiac mass and growth in both length and width. This process is fully reversible and has preserved or increased contractile function (Nakamura and Sadoshima, 2018). Initially induced as a compensatory response with concentric growth of the ventricle, pathological hypertrophy is irreversibly turned to ventricular chamber dilatation with wall thinning through lengthening of individual cardiomyocytes, contractile dysfunction, and heart failure (Schiattarella and Hill, 2015).
Strikingly, several lines of evidence suggest that the increased rate of cardiomyocyte proliferation is linked to physiological hypertrophy. The inhibition of C/EBPβ mediated by AKT1 increases CITED4 expression, stimulating cardiomyocyte proliferation and thus promoting physiological hypertrophy rather than pathological hypertrophy (Boström et al., 2010). However, not all experimental models showed similar results. For instance, the overexpression of Parbp1 stimulated cardiac physiology hypertrophy without increased proliferation (Baroldi et al., 1967). Understanding the link between physiological hypertrophy and proliferation will help stimulate CMs regeneration as well as maintain or improve cardiac function in patients with cardiovascular diseases. A growing body of evidence suggests that specific signaling mechanisms might be the primary determinants of hypertrophy types (Riehle et al., 2014; Rose et al., 2010; Takimoto et al., 2009; Tamirisa et al., 1999). For example, calcineurin dephosphorylates the nuclear factor of activated T cells (NFAT) and promotes NFAT nuclear localization. NFAT induces hypertrophy-related gene expression by interacting with transcriptional cofactors (Molkentin et al., 1998). Conversely, the calcineurin–NFAT pathway exerts no effect on physiological hypertrophy during exercise or pregnancy (Wilkins et al., 2004). It may be delightful to work toward the development of therapeutic approaches aimed at selectively inhibiting signaling mechanisms mediating pathological hypertrophy while preserving or even promoting those mediating physiological hypertrophy.
Nuclear polyploidy and multinucleation
DNA synthesis cannot be equal to cardiomyocyte proliferation, given that the process of polyploidization is typical in the adult heart (Beltrami et al., 2001; Broughton and Sussman, 2019; Hsieh et al., 2007). Mononucleated polyploid cells result from a cell duplicating its DNA during the S phase without undergoing nuclear division (Derks and Bergmann, 2020). Multinucleation in CMs is often the result of failed cytokinesis, concomitant with finished nuclear division (Liu et al., 2010). While nuclear polyploidy and multinucleation are common in mammalian ventricular cardiomyocytes, there is still very little understanding of whether polyploid and multinucleated CMs can undergo cell division, andthe related mechanisms. There is a consensus among nuclear ploidy and multinucleation that they have a proliferative advantage, thus inhibiting regeneration (Gan et al., 2020; Patterson et al., 2017; Patterson and Swift, 2019). The evidence reviewed here seems to suggest that therapeutic strategies remain narrow in focus dealing only with cardiomyocyte proliferation without polyploidy.
Polyploid CMs show great diversity among species. The zebrafish’s heart mainly contains diploid CMs that proliferate throughout life (González-Rosa et al., 2017). Human adult CMs mostly contain a single polyploid nucleus, while other mammalian species, such as rats and pigs, exhibit predominantly binucleated hearts (Alkass et al., 2015; Bergmann et al., 2015; Gräbner and Pfitzer, 1974). Considering that has been mentioned so far, it can be assumed that high ploidy levels in CMs are associated with the lost ability of the heart to regenerate. In addition, the ploidy levels appear to be related to cardiomyocyte types. While about 80% of CMs in the ventricles are binucleated, the proportion only accounts for 14% in mouse atria (Raulf et al., 2015). Several lines of evidence suggest that mononucleated cells have distinctive electrophysiological characteristics, including a high arrhythmogenicity (Huang et al., 2012); whether the ploidy level results from differences in metabolic activity and electrophysiological properties between cardiomyocytes remains unclear.
The increased cardiomyocyte ploidy is related to disease conditions. Pathological cardiac overload, such as hypertension, led to cardiomyocyte hypertrophy and heart enlargement and, at late stages, resulted in polyploidization in human hearts (Brodsky et al., 1994; Vliegen et al., 1995). Similar observations obviously apply to the mice (Liu et al., 2010). Following MI, except for compensatory hypertrophy in the remaining intact parts of the ventricles, most CMs with positive cell cycle marker Ki-67 in the peri-infarct area exhibited polyploid in human hearts (Meckert et al., 2005). Though cardiomyocyte hypertrophy accompanied by polyploidization is common in humans and mice, whether polyploidy is a prerequisite or a consequence of hypertrophic growth is unclear.
Amitosis
Amitosis is recognized as a specialized form of cell division with simple cleavage of the nucleus without the formation of a spindle figure or chromosomes. This process widely exists in prokaryotes and eukaryotes (Vitali et al., 2021), including hepatocytes (David and Uerlings, 1992), pancreatic acinar cells (Nagata, 2003), and macrophages of humans and other mammals (Arkhipov et al., 2008; Il'in et al., 2018; Iljine et al., 2013). So far, little attention has been paid to the role of amitosis in CM proliferation. In the 1980s, Pefitzer et.al proposed that the formation of binucleate myocardial cells and occasionally dumbbell-shaped restitution nuclei resulted from mitosis with impeded cellular divisions rather than amitosis (Pfitzer, 1980). However, binuclear cells gradually increased following the decreased mitotic cells during developmental stages. Guo et al., speculated that binuclear cells came from amitosis, which might be a primary division pattern during the adult stage (Guo et al., 2011). Shi and his colleagues also found that the goat CMs proliferated mainly in an amitotic way through nuclear transverse splitting by observing the paraffin sections of the hearts of 12 adult goats. These evident amitotic phases existed in adult goat ventricular muscle cells, including working and conducting cardiac muscle cells (Shi, 2007). Collectively, there still needs a more scientific understanding of amitosis in CM proliferation.
Regulation of miRNAs in CMs proliferation
Several of the miRNAs expressed in embryonic stem cells were reported to induce the proliferation of neonatal CMs. It is worth noting that these miRNAs share an identical or almost identical seed sequence, thus binding to a pile of overlapping targets of mRNAs. For instance, miR302-367 clusters were shown to decrease in expression during development and concomitant with the loss of CM proliferation capacity while they were reactivated after MI-induced cardiac proliferation (Tian et al., 2015). They were encoded by a single polycistronic transcript and produced five precursor miRNAs, including miR-302a, mir-302b, miR-302c, miR-302d, and the unrelated miR-367, the first of which shared the same seed sequence AAGUGCU (Barroso-del Jesus et al., 2009).
Recent advances have identified specific miRNA contributions to cardiac development and disease, as well as cardiomyocyte apoptosis and cell-cycle reentry after injury, which may generate fresh insights into developing miRNA diagnostics and miRNA inhibitors.
The role of specific miRNAs during cardiac development
Several published studies outline the critical role of miRNAs in myocardial development in the embryo (Fig. 2). miR-1 and miR-133 were the first identified miRNAs necessary for heart development, the former being the most abundant miRNA in the mouse heart (Zhao et al., 2007). While miR-1 and the related miR-133 arose from a common precursor RNA and worked together to promote the differentiation of mesoderm to CMs by targeting Dll-1, miR-1 stimulated, and miR-133 inhibited cardiac formation in the mid-late stage (Porrello, 2013). Serum response factor (SRF) acts as a regulator of cardiac and smooth muscle differentiation genes in cell cycle progression. The mesodermal gene expression of SRF-deficient embryonic stem cells could be promoted by miR-1 and miR-133 (Ivey et al., 2008). The positive effects of miR-1 in cardiac development are mainly mediated by targeting the IRX5 and Hand2 transcription factors (Ono et al., 2011). Conversely, the transcription factor NKX2-5 acted as an upstream inhibitor of miR-1 (Qian et al., 2011). Further, the overexpression of miR-1 mediated the inhibition of myocardial differentiation via targeting cdk9, which induced the activation of cardiac-specific genes (Takaya et al., 2009). In addition, cyclinD2, SRF3, and CDC42 were identified as targets of miR-133, inhibiting cardiomyocyte proliferation in the developing heart (Carè et al., 2007; Liu et al., 2008). Knock-down experiments confirmed that the loss of miR-133a led to lethal ventricular-septal defects in embryos (Liu et al., 2008).
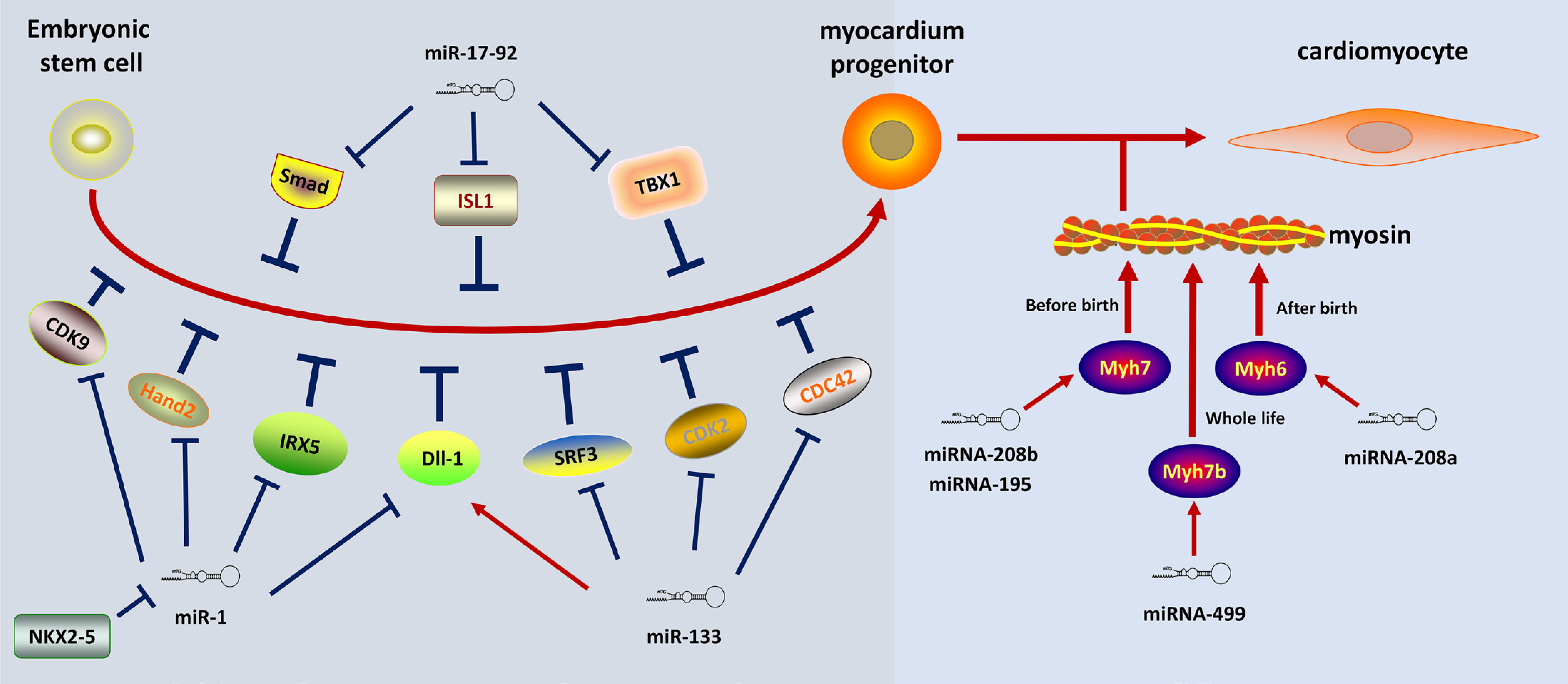
Fig. 2. The role of specific miRNAs during cardiac development
The red arrow shows the stimulatory modification while the blue “T” symbol represents the inhibitory modification.
The miR-17-92 cluster is essential for embryonic cardiac development and postnatal cardiogenesis stages. miR-17-92 cluster was indispensable for signaling cascade in the second heart field via targeting conserved Smad-binding sites, as required for the differentiation of the cardiac outflow tract (Wang et al., 2010). Genomic deletion of the miR-17-92 cluster led to ventricular-septum defects and lethal lung hypoplasia (Ventura et al., 2008). Additionally, the downregulation of the cardiac progenitor genes ISL1 and TBX1 appear to be under the control of miR-17-92, thus mediating myocardial differentiation (Wang et al., 2010).
The expression of myosin heavy chain protein of the adult heart was mediated by miR-499, miR-208a, and miR-208b, which expressed in parallel with their respective host genes during development. miR-208a resides in the myosin heavy chain 6 (Myh6) gene encoding the myosin heavy chain protein of the adult heart, whereas miR-208b and miR-499 are co-expressed together with the slow myosin Myh7 and Myh7b (Callis et al., 2009). Following the inhibition of miR-208a expression, fast skeletal markers were shown in the adult heart, suggesting the Myh6 intron-encoded miR-208a guided cardiac differentiation (van Rooij et al., 2007). Thrap1 (a component of the thyroid hormone receptor complex) and myostatin were identified as potential targets of miR-208, mediating effects on miR-208 in the developing and adult heart. Of note, miR-208a/b and miR-499 primarily play a surrogate role during heart development, suggesting the combination of miR-208 and Thrap1 might be nonessential for early cardiac development (Boettger and Braun, 2012). Additionally, miR-208a positively regulates the CX40, HOPX, and GATA4 genes, known as the powerful regulators of heart expression profile and function (Callis et al., 2009). miR-499 promotes the differentiation of cardiac progenitor into CMs via suppressing SOX6 (Pang et al., 2019). Further, upon initiating of cardiac hESC differentiation, miR-499 facilitates ventricular specification (Fu et al., 2011).
miR-195 and miR-497, identified as members of the miR-15 family, were upregulated during early postnatal cardiac development of the mice between postnatal day one and postnatal day ten. In a consistent manner, CMs exited from cell proliferation with subsequent binucleation. In contrast, it has also been shown that miR-195 overexpression in the embryonic heart inhibited the expression of mitotic genes and caused precocious cell cycle arrest (Porrello et al., 2011a).
Specific miRNAs in cell-cycle reentry after injury
Cardiomyocyte apoptosis is associated with cardiac disorders, including MI, cardiomyopathy, cardiac hypertrophy, and anthracycline-induced cardiotoxicity. The cardiomyocyte apoptosis cannot be compensated by efficient cell proliferation, thus resulting in contractile dysfunction and the progression of the disease. Recent advances in identifying miRNA that contributed to cardiomyocyte apoptosis may provide a mechanism for the pathogenesis after injury (Fig. 3A). Following ischemia/reperfusion, the expression of miR-320 was shown to increase via targeting heat-shock protein 20 (Hsp20) which is known as a cardioprotective protein. Further, knock-down experiments confirmed the defect of miR-320 can attenuate cell death after injury (Ren et al., 2009). Programmed cell death 4 (PDCD4) appears to be the primary target of miR-21, mediating the protective effects on the H2O2-induced injury. Additionally, Activator protein 1 (AP-1) was identified as the downstream signaling molecule of PDCD4 that is involved in the miR-21-mediated effect on cardiac myocytes (Cheng et al., 2009). The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis. miR-1 promotes apoptosis by downregulating heat shock protein-60 and heat shock protein-70 levels, while miR-133 participates in the inactivation of apoptosis by reducing caspase-9 expression (Xu et al., 2007). The hypoxia-inducible factor (Hif)-1α and Sirtuin1 (Sirt-1) were inhibited after the decrease of miR-199a, which recapitulates hypoxia preconditioning in CMs (Rane et al., 2009). The cardioprotective roles of miR-214 after injury were attributed to the repression of the mRNA encoding sodium/calcium exchanger 1 (Ncx1) to attenuate calcium overload, thus repressing several downstream effectors of Ca²⁺ signaling that mediated cell death. Moreover, miR-214 promoted cell survival by inhibiting the expression of the pro-apoptotic protein BIM (Aurora et al., 2012). Mitochondrial fission is involved in apoptosis initiation. miR-30 family members contributed to the inhibition of mitochondrial fission and apoptosis by suppressing the expression of p53, which upregulates dynamin-related protein-1 (Drp1) (Li et al., 2010). Considering all of this evidence, it seems that miRNAs are involved in cardiac apoptotic machinery, which might indeed be translatable to future approaches by inhibiting apoptosis after injury.
Following the decreased number of CMs induced by MI, the subject of the following might be focused on facilitating cell cycle re-entry of CMs by manipulating the miRNA network (Fig. 3B). Cyclin D2 and SRF were identified as primary targets of miR-133, mediating the regulation of the retinoblastoma protein phosphorylation during the G1 period (Liu et al., 2008). miRNA-590 and miRNA-119a promoted the cell cycle re-entry of adult CMs by downregulating HOPX, thus promoting the activity of cyclin D1/CDK4 complex and increasing cardiomyocyte proliferation in neonatal and adult animals. This process induced cardiac regeneration after MI based on the high-throughput screening by a whole-genome miRNA library for the significant miRNAs in the process of CMs proliferation (Eulalio et al., 2012). Moreover, the overexpression of miR-294 in adult mice hearts promoted the cell cycle re-entry of CMs by repressing the expression of Wee1, thus increasing the activity of cyclin B1/CDK1 complex (Borden et al., 2019). In addition, the miR-15 family miRNA bound to various components of the cell cycle and DNA damage response machinery, including the checkpoint kinase 1 (Chek1), whose inhibition led to improved cardiac repair after MI (Porrello et al., 2013). The cardiac-specific miR-128 regulated cell cycle controller expression by facilitating the expression of Cyclin E and CDK2 (the positive cell cycle regulators) through the Polycomb Repressive Complex-associated Suz12 protein (Huang et al., 2018).
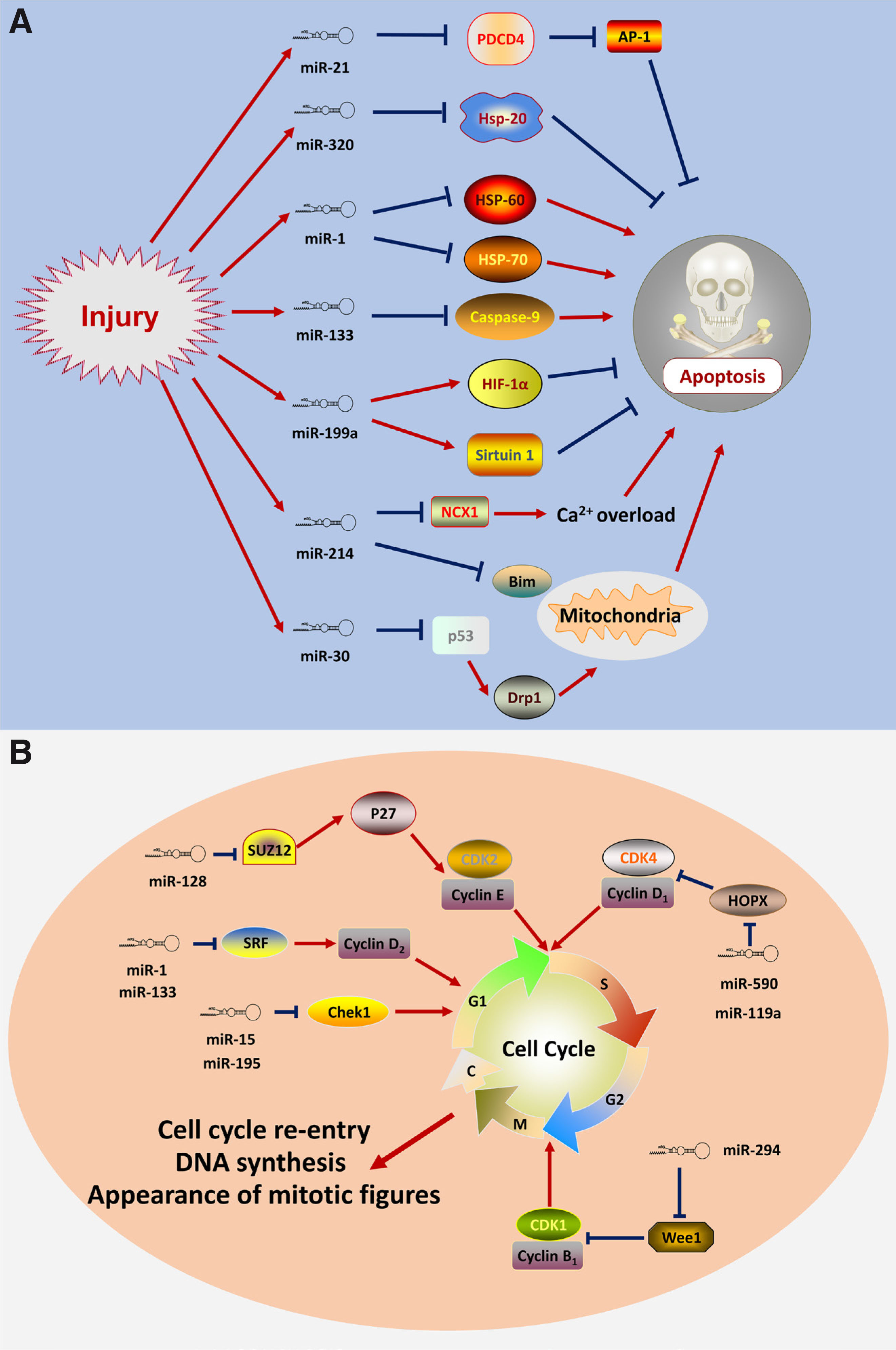
Fig. 3. Specific miRNAs in cell-cycle re-entry after injury
(A) MiRNAs affect the apoptosis process of cardiomyocytes after injury. (B) The cell cycle-relevant miRNAs regulate the cell cycle re-entry and DNA synthesis. The red arrow shows the stimulatory modification while the blue “T” symbol represents the inhibitory modification.
Discussion
Humans are not qualified to repair a “broken” heart now, whereas the zebrafish, regarded as the most classical model in heart development and regeneration, showed its excellent ability to regenerate (Sehring et al., 2016). There are remarkable differences in adult hearts between mammals and lower vertebrates in regeneration based onontogenetic or phylogenetic factors. Acquaintance with similarities in the trigger factors of cardiac regeneration between zebrafish and mammals may help us find more applications in mammals and therapeutic strategies.
The relevant therapeutic approaches have focused on the replacing of dead or dying CMs, for instance, by autologous cell transplantation of muscle progenitor cells or adult stem cells (Hirose et al., 2019), in parallel with unclear benefits, mode of action, and potential side effects. Bergmann et al., showed a high turnover rate of cardiac mesenchymal stem cells (CMSCs) (Bergmann et al., 2015), which signaled cardiomyocytes thus playing a crucial role in regeneration after injury. According to shared functions and commonly expressed biomarkers, Cencioni et al., referred to CMSCs as a general population, including cardiac precursor cells, pericytes, or cardiac fibroblasts (Chiara Cencioni et al., 2017). However, the latter cell population showed a limited differentiation potential under appropriate conditions (Driesen et al., 2014). CMSCs actively participated in the process of cardiac remodeling, emerging as a source of cardiac stem cell reservoir or secreting extracellular matrix to maintain the physical and mechanical integrity (Souders et al., 2009), whose therapeutic effects could also be enhanced by genetic and epigenetic interventions (Ieda et al., 2010; Jayawardena et al., 2015; Qian et al., 2012). CMSCs also contributed to the cardiac electrical conduction system based on the interconnection with cardiomyocyte gap junctions enriched in Cx43 and Cx45 (Zeigler et al., 2016). Additionally, cardiac fibroblasts, which account for a majority of cells in the heart, were reprogramed into functional cardiac-like myocytes with forced expression of four transcription factors, GATA4, HAND2, MEF2C, and TBX5, improving cardiac function and reducing adverse ventricular remodeling following myocardial infarction (Song et al., 2012). With the plasticity, reduced immunogenicity, and the relative simplicity of isolation and culture ex vivo (Gourdie et al., 2016), CMSCs become a strategy for the exogenous replacement of necrotic myocardium.
An alternative target is stimulating the re-entry of mature CM into the cell cycle. Increasing numbers of evidence showed that miRNA controlled this process both positively and negatively after MI. The miRNAs, which were not expressed in the heart but impacted cell proliferation by delivering to CMs exogenously, also had obvious therapeutic implications. Several human miRNAs were identified to induce CM proliferation, including CMs of neonatal rodents and human iPS-derived CMs (Eulalio et al., 2012). One of the most effective pro-proliferation miRNAs is miR-199a-3p. miR-199a-3p, known as a tumor suppressor, directly targeted the regulatory factors in cancer cell proliferation and metastasis, including c-Met (Migliore et al., 2008), mammalian target of rapamycin (Callegari et al., 2018) and CD44 (Gao et al., 2015). In CMs, miR-199a-3p expressed at a relatively low level after birth while its exogenous transaction remarkably induced CMs proliferation in both neonatal and adult heart (Pandey and Ahmed, 2015), lighting on exploring potential therapeutic approaches in humans. The let-7 family, belonging to inhibitory miRNAs, were endogenously expressed in CMs, especially in the transition from fetal to neonatal life, and were highly expressed as CMs exited the cell cycle (Johnson et al., 2007). Invariably, the levels of these miRNA clusters were increased during heart regeneration in zebrafish compared to mammals in which regeneration failed to occur (Aguirre et al., 2014). The regenerative effects in mice by inhibiting these miRNAs after MI light on exploring potential therapeutic approaches of let-7 family miRNAs (Hu et al., 2019). Other inhibitory miRNAs included the muscle tissue-specific miR-1~133 cluster (Zhao et al., 2007; Zhao et al., 2005), particularly miR-133 by inhibiting cardiac regeneration in zebrafish and miR-128 by regulating cell cycle controller expression (Huang et al., 2018; Yin et al., 2012). Altogether, the observations reported so far hint at the possibility of achieving cardiac regeneration by delivering several miRNAs to CMs exogenously or inhibiting other miRNAs from re-entering the cell cycle. Despite the emergence of the engineered oligonucleotides called “antagomirs” to silence endogenous miRNAs (Lin et al., 2009), there remains a paucity of evidence on a rigorous assessment of the safety and improvement in delivery methods required for setting RNA-based therapeutics as an innovative reliable tool to promote cardiac regeneration in patients.
In addition to cardiomyocytes, many noncardiomyocyte types contribute to the maintenance of cardiac function and regeneration after injury with therapeutic potential. The epicardiumalso emerges as a source of vascular smooth muscle cells, pericytes, and fibroblasts during heart development and repair. It secretes essential factors for cardiomyocyte proliferation and survival and, therefore, represents an important target for therapeutic interventions (Packer, 2018). Macrophage recruitment is essential for heart regeneration through the secretion of oncostatin M (Li et al., 2020b). Additionally, monocular cells resident in the heart and T-regulatory cells during pregnancy played a paracrine regulatory role in CMs proliferation (Lavine et al., 2014; Zacchigna et al., 2018). The regulatory T-cells serve as a source of regenerative factors in a paracrine manner. Their secreted factors, such as chemokine ligand 24, growth arrest-specific 6, or amphiregulin, facilitate CMs proliferation (Li et al., 2019). Subsequently, Li et al., further demonstrated that the specific ablation of CD4+ T-cells potentiated heart regeneration and directly or indirectly facilitated the polarization of macrophages away from the pro-fibrotic M2-like signature in the juvenile heart (Li et al., 2020a).
The stroma signals to CMs, thus playing a crucial role in their proliferation. Lysophosphatidic acid (LPA) acts as a potent extracellular signal in various biological processes, and diseases and LPA3-mediated LPA signaling was shown to play a pivotal role in CMs proliferation (Wang et al., 2020a). The extracellular matrix component was shown to affect the growth and differentiation of CMs in mice, mediated by agrin (Bassat et al., 2017) and myeloid-derived growth factor (Wang et al., 2020b). The extracellular protein reelin (RELN) was required for efficient heart repair and function after neonatal MI. The exogenous delivery of RELN potentiated heart function in adult mice after MI (Liu et al., 2020). Work carried out in the past few years has explored a set of cytokines and growth factors known to stimulate the proliferation of neonatal CMs, including interleukin-6 (Przybyt et al., 2013), follistatin like 1 (Wei et al., 2015), platelet-derived growth factor (Hinrichsen et al., 2007), fibroblast growth factor family (Engel et al., 2006) and neuregulin-1 (Zhao et al., 1998).
Conclusion
Heart regeneration needs not only the proliferation of a single cell population but also the synergetic effects of different types of cells and extracellular matrix components. Although a growing body of evidence indicates the potential capacity of proliferation in the embryonic and neonatal heart by manipulating the miRNA network that is naturally permissive for regeneration, in parallel, there are less research focused on the stimulation of adult CM proliferation in adult or aging animal hearts. Hopefully, this review will contribute to a deeper understanding of heart regeneration in mammals and its microRNAs regulatory mechanisms that merit further and translational research to possibly one day be able to regenerate a diseased adult human heart.
Abbreviations
CMs, cardiomyocytes ; MI, myocardial infarction ; miRNA, microRNA ; MADM, Mosaic Analysis with Double Markers ; RISC, RNA-induced silencing complex ; 3'-UTR, 3' untranslated regions ; SRF, serum response factor ; FSTL1, follistatin-like 1 ; PDGFs, platelet-derived growth factors ; IGF, insulin-like growth factor ; ECM, extracellular matrix ; MMPs, matrix metallopeptidases ; IGF-1, insulin-like growth factor-1 ; AT1, angiotensin II type 1 receptor ; SV40, SV40 large T antigen ; pri-miRNA, primary miRNA ; pre-miRNA, precursor miRNAs ; AGO2, Argonaute proteins ; lncRNAs, long noncoding RNAs ; 14C, carbon-14 ; CDK1, cyclin-dependent kinase 1 ; Myh6, myosin heavy chain 6 ; Myh7, myosin heavy chain 7 ; Myh7b, myosin heavy chain 7b ; SRF, Serum response factor ; PDCD4, Programmed cell death 4 ; AP-1, Activator protein 1 ; Drp1, dynamin-related protein-1 ;Declarations
Conflicts of interest
The authors declare no conflict of interest associated with this manuscript.
Author contributions
Shu-ning Guo and Jie-han Li performed the literature search and drafted the manuscript. Both of them contributed equally to this review. Yu-rong Chai, Yi Ding, and Jing Li critically revised the work. All authors read and approved the final manuscript.
Funding
This research was supported by the Key Scientific Research Project of Higher Education Institution in Henan Province (No: 21A310022).
References
Aguirre A., Montserrat N., Zacchigna S., Nivet E., Hishida T., Krause M. N., Kurian L., Ocampo A., Vazquez-Ferrer E., Rodriguez-Esteban C., Kumar S., Moresco J. J., Yates J. R., Campistol J. M., Sancho-Martinez I., Giacca M., Izpisua Belmonte J. C. (2014). In Vivo Activation of a Conserved MicroRNA Program Induces Mammalian Heart Regeneration. Cell Stem Cell 15: 589-604.
Ali S. R., Hippenmeyer S., Saadat L. V., Luo L., Weissman I. L., Ardehali R. (2014). Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proceedings of the National Academy of Sciences 111: 8850-8855.
Alkass K., Panula J., Westman M., Wu T.D., Guerquin-Kern J.L., Bergmann O. (2015). No Evidence for Cardiomyocyte Number Expansion in Preadolescent Mice. Cell 163: 1026-1036.
Ameres S. L., Zamore P. D. (2013). Diversifying microRNA sequence and function. Nature Reviews Molecular Cell Biology 14: 475-488.
Arkhipov S. A., Shkurupiy V. A., Ijine D. A., Ignatovich N. V., Akhromenko E. S., Arkhipova V. V. (2008). Formation and Some Cytophysiological Characteristics of Polynuclear Macrophages in Primary Cultures of Peritoneal Cells. Bulletin of Experimental Biology and Medicine 146: 838-841.
Aurora A. B., Mahmoud A. I., Luo X., Johnson B. A., van Rooij E., Matsuzaki S., Humphries K. M., Hill J. A., Bassel-Duby R., Sadek H. A., Olson E. N. (2012). MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca2+ overload and cell death. Journal of Clinical Investigation 122: 1222-1232.
Bae J., Salamon R. J., Brandt E. B., Paltzer W. G., Zhang Z., Britt E. C., Hacker T. A., Fan J., Mahmoud A. I. (2021). Malonate Promotes Adult Cardiomyocyte Proliferation and Heart Regeneration. Circulation 143: 1973-1986.
Baehr A., Umansky K. B., Bassat E., Jurisch V., Klett K., Bozoglu T., Hornaschewitz N., Solyanik O., Kain D., Ferraro B., Cohen-Rabi R., Krane M., Cyran C., Soehnlein O., Laugwitz K. L., Hinkel R., Kupatt C., Tzahor E. (2020). Agrin Promotes Coordinated Therapeutic Processes Leading to Improved Cardiac Repair in Pigs. Circulation 142: 868-881.
Baidyuk E. V., Sakuta G. A., Vorobev M. L., Stepanov A. V., Karpov A. A., Rogoza O. V., Kudryavtsev B. N. (2019). Rat Left Ventricular Cardiomyocytes Characterization in the Process of Postinfarction Myocardial Remodeling. Cytometry Part A 95: 730-736.
Baroldi G., Falzi G., Lampertico P. (1967). The Nuclear Patterns of the Cardiac Muscle Fiber. Cardiology 51: 109-123.
Barroso-del Jesus A., Lucena-Aguilar G., Menendez P. (2009). The miR-302-367 cluster as a potential stemness regulator in ESCs. Cell Cycle 8: 394-398.
Bassat E., Mutlak Y. E., Genzelinakh A., Shadrin I. Y., Baruch Umansky K., Yifa O., Kain D., Rajchman D., Leach J., Riabov Bassat D., Udi Y., Sarig R., Sagi I., Martin J. F., Bursac N., Cohen S., Tzahor E. (2017). The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 547: 179-184.
Beltrami A. P., Urbanek K., Kajstura J., Yan S.M., Finato N., Bussani R., Nadal-Ginard B., Silvestri F., Leri A., Beltrami C. A., Anversa P. (2001). Evidence That Human Cardiac Myocytes Divide after Myocardial Infarction. New England Journal of Medicine 344: 1750-1757.
Bergmann O., Bhardwaj R. D., Bernard S., Zdunek S., Barnabé-Heider F., Walsh S., Zupicich J., Alkass K., Buchholz B. A., Druid H., Jovinge S., Frisén J. (2009). Evidence for Cardiomyocyte Renewal in Humans. Science 324: 98-102.
Bergmann O., Zdunek S., Felker A., Salehpour M., Alkass K., Bernard S., Sjostrom S. L., Szewczykowska M., Jackowska T., dos Remedios C., Malm T., Andrä M., Jashari R., Nyengaard J. R., Possnert G., Jovinge S., Druid H., Frisén J. (2015). Dynamics of Cell Generation and Turnover in the Human Heart. Cell 161: 1566-1575.
Bettex D. A., Pretre R., Chassot P.G. (2014). Is our heart a well-designed pump? The heart along animal evolution. European Heart Journal 35: 2322-2332.
Boettger T., Braun T. (2012). A New Level of Complexity. Circulation Research 110: 1000-1013.
Borden A., Kurian J., Nickoloff E., Yang Y., Troupes C. D., Ibetti J., Lucchese A. M., Gao E., Mohsin S., Koch W. J., Houser S. R., Kishore R., Khan M. (2019). Transient Introduction of miR-294 in the Heart Promotes Cardiomyocyte Cell Cycle Reentry After Injury. Circulation Research 125: 14-25.
Boström P., Mann N., Wu J., Quintero P. A., Plovie E. R., Panáková D., Gupta R. K., Xiao C., MacRae C. A., Rosenzweig A., Spiegelman B. M. (2010). C/EBPβ Controls Exercise-Induced Cardiac Growth and Protects against Pathological Cardiac Remodeling. Cell 143: 1072-1083.
Boulton J., HENRY R., Roddick L. G., Rogers D., Thompson L., Warner G. (1991). Survival After Neonatal Myocardial Infarction. Pediatrics 88: 145-150.
Brodsky V.Y., Arefyeva A.M., Gvasava I.G., Sarkisov D.S., Panova N.W. (1994). Polyploidy in cardiac myocytes of normal and hypertrophic human hearts; range of values. Virchows Archiv 424: 429-435.
Broughton K. M., Sussman M. A. (2019). Adult Cardiomyocyte Cell Cycle Detour: Off-ramp to Quiescent Destinations. Trends in Endocrinology & Metabolism 30: 557-567.
Callegari E., D’Abundo L., Guerriero P., Simioni C., Elamin B. K., Russo M., Cani A., Bassi C., Zagatti B., Giacomelli L., Blandamura S., Moshiri F., Ultimo S., Frassoldati A., Altavilla G., Gramantieri L., Neri L. M., Sabbioni S., Negrini M. (2018). miR-199a-3p Modulates MTOR and PAK4 Pathways and Inhibits Tumor Growth in a Hepatocellular Carcinoma Transgenic Mouse Model. Molecular Therapy - Nucleic Acids 11: 485-493.
Callis T. E., Pandya K., Seok H. Y., Tang R.H., Tatsuguchi M., Huang Z.P., Chen J.F., Deng Z., Gunn B., Shumate J., Willis M. S., Selzman C. H., Wang D.Z. (2009). MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. Journal of Clinical Investigation 119: 2772-2786.
Camacho P., Fan H., Liu Z., He J.Q. (2016). Large Mammalian Animal Models of Heart Disease. Journal of Cardiovascular Development and Disease 3: 30.
Cardoso A. C., Lam N. T., Savla J. J., Nakada Y., Pereira A. H. M., Elnwasany A., Menendez-Montes I., Ensley E. L., Bezan Petric U., Sharma G., Sherry A. D., Malloy C. R., Khemtong C., Kinter M. T., Tan W. L. W., Anene-Nzelu C. G., Foo R. S.Y., Nguyen N. U. N., Li S., Ahmed M. S., Elhelaly W. M., Abdisalaam S., Asaithamby A., Xing C., Kanchwala M., Vale G., Eckert K. M., Mitsche M. A., McDonald J. G., Hill J. A., Huang L., Shaul P. W., Szweda L. I., Sadek H. A. (2020). Mitochondrial substrate utilization regulates cardiomyocyte cell-cycle progression. Nature Metabolism 2: 167-178.
Carè A., Catalucci D., Felicetti F., Bonci D., Addario A., Gallo P., Bang M.L., Segnalini P., Gu Y., Dalton N. D., Elia L., Latronico M. V. G., Høydal M., Autore C., Russo M. A., Dorn G. W., Ellingsen , Ruiz-Lozano P., Peterson K. L., Croce C. M., Peschle C., Condorelli G. (2007). MicroRNA-133 controls cardiac hypertrophy. Nature Medicine 13: 613-618.
Cesna S., Eicken A., Juenger H., Hess J. (2013). Successful Treatment of a Newborn With Acute Myocardial Infarction on the First Day of Life. Pediatric Cardiology 34: 1868-1870.
Chekulaeva M., Filipowicz W. (2009). Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Current Opinion in Cell Biology 21: 452-460.
Cheng Y., Liu X., Zhang S., Lin Y., Yang J., Zhang C. (2009). MicroRNA-21 protects against the H2O2-induced injury on cardiac myocytes via its target gene PDCD4. Journal of Molecular and Cellular Cardiology 47: 5-14.
Cencioni C., Atlante S., Savoia M., Martelli F., Farsetti A., Capogrossi M. C., Zeiher A. M., Gaetano C., Spallotta F. (2017). The double life of cardiac mesenchymal cells: Epimetabolic sensors and therapeutic assets for heart regeneration. Pharmacology & Therapeutics 171: 43-55.
Crick S. J., Sheppard M. N., Ho S., Gebstein L., Anderson R. H. (1998). Anatomy of the pig heart: comparisons with normal human cardiac structure. Journal of Anatomy 193: 105-119.
Cui M., Atmanli A., Morales M. G., Tan W., Chen K., Xiao X., Xu L., Liu N., Bassel-Duby R., Olson E. N. (2021). Nrf1 promotes heart regeneration and repair by regulating proteostasis and redox balance. Nature Communications 12: 5270.
David H., Uerlings I. (1992). Ultrastructure of amitosis and mitosis of the liver. Zentralblatt fur Pathologie 138: 278-283.
Derks W., Bergmann O. (2020). Polyploidy in Cardiomyocytes. Circulation Research 126: 552-565.
Deutsch M.A., Cleuziou J., Noebauer C., Eicken A., Vogt M., Hoerer J., Lange R., Schreiber C. (2014). Successful Management of Neonatal Myocardial Infarction with ECMO and Intracoronary r-tPA lysis. Congenital Heart Disease 9: E169-E174.
Drenckhahn J.D., Schwarz Q. P., Gray S., Laskowski A., Kiriazis H., Ming Z., Harvey R. P., Du X.J., Thorburn D. R., Cox T. C. (2008). Compensatory Growth of Healthy Cardiac Cells in the Presence of Diseased Cells Restores Tissue Homeostasis during Heart Development. Developmental Cell 15: 521-533.
Driesen R. B., Nagaraju C. K., Abi-Char J., Coenen T., Lijnen P. J., Fagard R. H., Sipido K. R., Petrov V. V. (2014). Reversible and irreversible differentiation of cardiac fibroblasts. Cardiovascular Research 101: 411-422.
Ebelt H., Hufnagel N., Neuhaus P., Neuhaus H., Gajawada P., Simm A., Müller-Werdan U., Werdan K., Braun T. (2005). Divergent Siblings. Circulation Research 96: 509-517.
Engel F. B., Hsieh P. C. H., Lee R. T., Keating M. T. (2006). FGF1/p38 MAP kinase inhibitor therapy induces cardiomyocyte mitosis, reduces scarring, and rescues function after myocardial infarction. Proceedings of the National Academy of Sciences 103: 15546-15551.
Eulalio A., Mano M., Ferro M. D., Zentilin L., Sinagra G., Zacchigna S., Giacca M. (2012). Functional screening identifies miRNAs inducing cardiac regeneration. Nature 492: 376-381.
Farooqi K. M., Sutton N., Weinstein S., Menegus M., Spindola-Franco H., Pass R. H. (2012). Neonatal Myocardial Infarction: Case Report and Review of the Literature. Congenital Heart Disease 7: E97-E102.
Fratz S., Hager A., Schreiber C., Schwaiger M., Hess J., Stern H. C. (2011). Long-Term Myocardial Scarring After Operation for Anomalous Left Coronary Artery From the Pulmonary Artery. The Annals of Thoracic Surgery 92: 1761-1765.
Fu J.D., Rushing S. N., Lieu D. K., Chan C. W., Kong C.W., Geng L., Wilson K. D., Chiamvimonvat N., Boheler K. R., Wu J. C., Keller G., Hajjar R. J., Li R. A. (2011). Distinct Roles of MicroRNA-1 and -499 in Ventricular Specification and Functional Maturation of Human Embryonic Stem Cell-Derived Cardiomyocytes. PLoS ONE 6: e27417.
Gabisonia K., Prosdocimo G., Aquaro G. D., Carlucci L., Zentilin L., Secco I., Ali H., Braga L., Gorgodze N., Bernini F., Burchielli S., Collesi C., Zandonà L., Sinagra G., Piacenti M., Zacchigna S., Bussani R., Recchia F. A., Giacca M. (2019). MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature 569: 418-422.
Gallet R., Dawkins J., Valle J., Simsolo E., de Couto G., Middleton R., Tseliou E., Luthringer D., Kreke M., Smith R. R., Marbán L., Ghaleh B., Marbán E. (2017). Exosomes secreted by cardiosphere-derived cells reduce scarring, attenuate adverse remodelling, and improve function in acute and chronic porcine myocardial infarction. European Heart Journal 38: 201-211.
Gamba L., Harrison M., Lien C.L. (2014). Cardiac Regeneration in Model Organisms. Current Treatment Options in Cardiovascular Medicine 16: 288.
Gan P., Patterson M., Sucov H. M. (2020). Cardiomyocyte Polyploidy and Implications for Heart Regeneration. Annual Review of Physiology 82: 45-61.
Gao Y., Feng Y., Shen J. K., Lin M., Choy E., Cote G. M., Harmon D. C., Mankin H. J., Hornicek F. J., Duan Z. (2015). CD44 is a direct target of miR-199a-3p and contributes to aggressive progression in osteosarcoma. Scientific Reports 5: 11365.
Genge C. E., Lin E., Lee L., Sheng X.Y., Rayani K., Gunawan M., Stevens C. M., Li A. Y., Talab S. S., Claydon T. W., Hove-Madsen L., Tibbits G. F. (2016). The Zebrafish Heart as a Model of Mammalian Cardiac Function. In Reviews of Physiology, Biochemistry and Pharmacology, Vol. 171. (Ed. Nilius Bernd, de Tombe Pieter, Gudermann Thomas, Jahn Reinhard, Lill Roland, Petersen Ole H.) Springer International Publishing, Cham.
González-Rosa J. M., Burns C. E., Burns C. G. (2017). Zebrafish heart regeneration: 15 years of discoveries. Regeneration 4: 105-123.
Gourdie R. G., Dimmeler S., Kohl P. (2016). Novel therapeutic strategies targeting fibroblasts and fibrosis in heart disease. Nature Reviews Drug Discovery 15: 620-638.
Gräbner W., Pfitzer P. (1974). Number of nuclei in isolated myocardial cells of pigs. Virchows Archiv B Cell Pathology 15: 279-294.
Guo Z., Lin J., Chang L., Xu Z., Cai X. (2011). Features of Cardiomyocyte Division During Rat Heart Development. Pakistan Journal of Zoology 43: 321-330.
Haubner B. J., Adamowicz-Brice M., Khadayate S., Tiefenthaler V., Metzler B., Aitman T., Penninger J. M. (2012). Complete cardiac regeneration in a mouse model of myocardial infarction. Aging 4: 966-977.
Haubner B. J., Schneider J., Schweigmann U., Schuetz T., Dichtl W., Velik-Salchner C., Stein J.I., Penninger J. M. (2016). Functional Recovery of a Human Neonatal Heart After Severe Myocardial Infarction. Circulation Research 118: 216-221.
Hinrichsen R., HaunsØ S., Hinrichsen R., HaunsØ S., Busk P. K., Hinrichsen R., HaunsØ S., Busk P. K. (2007). Different regulation of p27 and Akt during cardiomyocyte proliferation and hypertrophy. Growth Factors 25: 132-140.
Hirose K., Payumo A. Y., Cutie S., Hoang A., Zhang H., Guyot R., Lunn D., Bigley R. B., Yu H., Wang J., Smith M., Gillett E., Muroy S. E., Schmid T., Wilson E., Field K. A., Reeder D.A. M., Maden M., Yartsev M. M., Wolfgang M. J., Grützner F., Scanlan T. S., Szweda L. I., Buffenstein R., Hu G., Flamant F., Olgin J. E., Huang G. N. (2019). Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science 364: 184-188.
Hsieh P. C. H., Segers V. F. M., Davis M. E., MacGillivray C., Gannon J., Molkentin J. D., Robbins J., Lee R. T. (2007). Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nature Medicine 13: 970-974.
Hu Y., Jin G., Li B., Chen Y., Zhong L., Chen G., Chen X., Zhong J., Liao W., Liao Y., Wang Y., Bin J. (2019). Suppression of miRNA let-7i-5p promotes cardiomyocyte proliferation and repairs heart function post injury by targetting CCND2 and E2F2. Clinical Science 133: 425-441.
Huang C.F., Chen Y.C., Yeh H.I., Chen S.A. (2012). Mononucleated and binucleated cardiomyocytes in left atrium and pulmonary vein have different electrical activity and calcium dynamics. Progress in Biophysics and Molecular Biology 108: 64-73.
Huang W., Feng Y., Liang J., Yu H., Wang C., Wang B., Wang M., Jiang L., Meng W., Cai W., Medvedovic M., Chen J., Paul C., Davidson W. S., Sadayappan S., Stambrook P. J., Yu X.Y., Wang Y. (2018). Loss of microRNA-128 promotes cardiomyocyte proliferation and heart regeneration. Nature Communications 9: 700.
Ieda M., Fu J.D., Delgado-Olguin P., Vedantham V., Hayashi Y., Bruneau B. G., Srivastava D. (2010). Direct Reprogramming of Fibroblasts into Functional Cardiomyocytes by Defined Factors. Cell 142: 375-386.
Il’in D. A., Arkhipov S. A., Shkurupy V. A. (2018). Analysis of IL-1α, bFGF, TGF-β1, IFNγ, MMP-1, and CatD Expression in Multinuclea Macrophages In Vitro. Bulletin of Experimental Biology and Medicine 164: 456-458.
Iljine D. A., Arkhipov S. A., Shkurupy V. A. (2013). In Vitro Expression of IL-1α, GM-CSF, and TNF-α by Multinucleated Macrophages from BCG-Infected Mice. Bulletin of Experimental Biology and Medicine 155: 663-666.
Ivey K. N., Muth A., Arnold J., King F. W., Yeh R.F., Fish J. E., Hsiao E. C., Schwartz R. J., Conklin B. R., Bernstein H. S., Srivastava D. (2008). MicroRNA Regulation of Cell Lineages in Mouse and Human Embryonic Stem Cells. Cell Stem Cell 2: 219-229.
Jayawardena T. M., Finch E. A., Zhang L., Zhang H., Hodgkinson C. P., Pratt R. E., Rosenberg P. B., Mirotsou M., Dzau V. J. (2015). MicroRNA Induced Cardiac Reprogramming In Vivo. Circulation Research 116: 418-424.
Johnson C. D., Esquela-Kerscher A., Stefani G., Byrom M., Kelnar K., Ovcharenko D., Wilson M., Wang X., Shelton J., Shingara J., Chin L., Brown D., Slack F. J. (2007). The let-7 MicroRNA Represses Cell Proliferation Pathways in Human Cells . Cancer Research 67: 7713-7722.
Lagos-Quintana M., Rauhut R., Lendeckel W., Tuschl T. (2001). Identification of Novel Genes Coding for Small Expressed RNAs. Science 294: 853-858.
Lavine K. J., Epelman S., Uchida K., Weber K. J., Nichols C. G., Schilling J. D., Ornitz D. M., Randolph G. J., Mann D. L. (2014). Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proceedings of the National Academy of Sciences 111: 16029-16034.
Levin I., Naegler T., Kromer B., Diehl M., Francey R. J., Gomez-Pelaez A. J., Steele L. P., Wagenbach D., Weller R., Worthy D. E. (2010). Observations and modelling of the global distribution and long-term trend of atmospheric <sup>14</sup>CO<sub>2</sub>. Tellus B: Chemical and Physical Meteorology 62: 26.
Levy P. T., Tissot C., Horsberg Eriksen B., Nestaas E., Rogerson S., McNamara P. J., El-Khuffash A., de Boode W. P., Austin T., Bohlin K., Bravo M. C., Breatnach C. R., Breindahl M., Dempsey E., Groves A. M., Gupta S., Molnar Z., Roehr C. C., Savoia M., Schubert U., Schwarz C. E., Sehgal A., Singh Y., Slieker M. G., van der Lee R., van Laere D., van Overmeire B., van Wyk L. (2018). Application of Neonatologist Performed Echocardiography in the Assessment and Management of Neonatal Heart Failure unrelated to Congenital Heart Disease. Pediatric Research 84: 78-88.
Li J., Donath S., Li Y., Qin D., Prabhakar B. S., Li P. (2010). Correction: miR-30 Regulates Mitochondrial Fission through Targeting p53 and the Dynamin-Related Protein-1 Pathway. PLoS Genetics 6: 1000795.
Li J., Guo S., Sun Z., Fu Y. (2022). Noncoding RNAs in Drug Resistance of Gastrointestinal Stromal Tumor. Frontiers in Cell and Developmental Biology 10: 808591.
Li J., Liang C., Yang K. Y., Huang X., Han M. Y., Li X., Chan V. W., Chan K. S., Liu D., Huang Z.P., Zhou B., Lui K. O. (2020a). Specific ablation of CD4 + T-cells promotes heart regeneration in juvenile mice . Theranostics 10: 8018-8035.
Li J., Yang K. Y., Tam R. C. Y., Chan V. W., Lan H. Y., Hori S., Zhou B., Lui K. O. (2019). Regulatory T-cells regulate neonatal heart regeneration by potentiating cardiomyocyte proliferation in a paracrine manner. Theranostics 9: 4324-4341.
Li Y., Feng J., Song S., Li H., Yang H., Zhou B., Li Y., Yue Z., Lian H., Liu L., Hu S., Nie Y. (2020b). gp130 Controls Cardiomyocyte Proliferation and Heart Regeneration. Circulation 142: 967-982.
Lin Z., Murtaza I., Wang K., Jiao J., Gao J., Li P.F. (2009). miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proceedings of the National Academy of Sciences 106: 12103-12108.
Liu N., Bezprozvannaya S., Williams A. H., Qi X., Richardson J. A., Bassel-Duby R., Olson E. N. (2008). microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes & Development 22: 3242-3254.
Liu X., De la Cruz E., Gu X., Balint L., Oxendine-Burns M., Terrones T., Ma W., Kuo H.H., Lantz C., Bansal T., Thorp E., Burridge P., Jakus Z., Herz J., Cleaver O., Torres M., Oliver G. (2020). Lymphoangiocrine signals promote cardiac growth and repair. Nature 588: 705-711.
Liu Z., Yue S., Chen X., Kubin T., Braun T. (2010). Regulation of Cardiomyocyte Polyploidy and Multinucleation by CyclinG1. Circulation Research 106: 1498-1506.
Mackintosh N., Armstrong N. (2020). Understanding and managing uncertainty in health care: revisiting and advancing sociological contributions. Sociology of Health & Illness 42: 1-20.
Mahmoud A. I., Kocabas F., Muralidhar S. A., Kimura W., Koura A. S., Thet S., Porrello E. R., Sadek H. A. (2013). Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 497: 249-253.
Meckert P., Rivello H., Vigliano C., GonzaleZ P., Favaloro R., Laguens R. (2005). Endomitosis and polyploidization of myocardial cells in the periphery of human acute myocardial infarction. Cardiovascular Research 67: 116-123.
Migliore C., Petrelli A., Ghiso E., Corso S., Capparuccia L., Eramo A., Comoglio P. M., Giordano S. (2008). MicroRNAs Impair MET-Mediated Invasive Growth. Cancer Research 68: 10128-10136.
Mohamed T. M.A., Ang Y.S., Radzinsky E., Zhou P., Huang Y., Elfenbein A., Foley A., Magnitsky S., Srivastava D. (2018). Regulation of Cell Cycle to Stimulate Adult Cardiomyocyte Proliferation and Cardiac Regeneration. Cell 173: 104-116.e12.
Molkentin J. D., Lu J.R., Antos C. L., Markham B., Richardson J., Robbins J., Grant S. R., Olson E. N. (1998). A Calcineurin-Dependent Transcriptional Pathway for Cardiac Hypertrophy. Cell 93: 215-228.
Mollova M., Bersell K., Walsh S., Savla J., Das L. T., Park S.Y., Silberstein L. E., dos Remedios C. G., Graham D., Colan S., Kühn B. (2013). Cardiomyocyte proliferation contributes to heart growth in young humans. Proceedings of the National Academy of Sciences 110: 1446-1451.
Murry C. E., Reinecke H., Pabon L. M. (2006). Regeneration Gaps. Journal of the American College of Cardiology 47: 1777-1785.
Murugan S. J., Gnanapragasam J., Vettukattil J. (2002). Acute myocardial infarction in the neonatal period. Cardiology in the Young 12: 411-413.
Nagata T., (2003). Light and electron microscopic radioautographic studies on macromolecular synthesis in amitotic hepatocytes of aging mice. Cellular and molecular biology (Noisy-le-Grand, France 49: 591-611.
Nakada Y., Canseco D. C., Thet S.W., Abdisalaam S., Asaithamby A., Santos C. X., Shah A. M., Zhang H., Faber J. E., Kinter M. T., Szweda L. I., Xing C., Hu Z., Deberardinis R. J., Schiattarella G., Hill J. A., Oz O., Lu Z., Zhang C. C., Kimura W., Sadek H. A. (2017). Hypoxia induces heart regeneration in adult mice. Nature 541: 222-227.
Nakada Y., Kimura W., Sadek H. A. (2015). Defining the Limit of Embryonic Heart Regeneration. Circulation 132: 77-78.
Nakamura M., Sadoshima J. (2018). Mechanisms of physiological and pathological cardiac hypertrophy. Nature Reviews Cardiology 15: 387-407.
Olivetti G., Melissari M., Capasso J. M., Anversa P. (1991). Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy.. Circulation Research 68: 1560-1568.
Ono K., Kuwabara Y., Han J. (2011). MicroRNAs and cardiovascular diseases. FEBS Journal 278: 1619-1633.
Packer M. (2018). Epicardial Adipose Tissue May Mediate Deleterious Effects of Obesity and Inflammation on the Myocardium. Journal of the American College of Cardiology 71: 2360-2372.
Pandey R., Ahmed R. P. (2015). MicroRNAs Inducing Proliferation of Quiescent Adult Cardiomyocytes. Cardiovascular regenerative medicine 2: e519.
Pang J. K. S., Phua Q. H., Soh B.S. (2019). Applications of miRNAs in cardiac development, disease progression and regeneration. Stem Cell Research & Therapy 10: 336.
Patterson M., Barske L., Van Handel B., Rau C. D., Gan P., Sharma A., Parikh S., Denholtz M., Huang Y., Yamaguchi Y., Shen H., Allayee H., Crump J. G., Force T. I., Lien C.L., Makita T., Lusis A. J., Kumar S. R., Sucov H. M. (2017). Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nature Genetics 49: 1346-1353.
Patterson M., Swift S. K. (2019). Residual Diploidy in Polyploid Tissues: A Cellular State with Enhanced Proliferative Capacity for Tissue Regeneration?. Stem Cells and Development 28: 1527-1539.
Peeters S., Vandenplas Y., Jochmans K., Bougatef A., Waele M. D., Wolf D. D. (1993). Myocardial infarction in a neonate with hereditary antithrombin III deficiency. Acta Paediatrica 82: 610-613.
Pfitzer P. (1980). Amitosis: A Historical Misinterpretation?. Pathology - Research and Practice 167: 292-300.
Polizzotti B. D., Ganapathy B., Haubner B. J., Penninger J. M., Kühn B. (2016). A cryoinjury model in neonatal mice for cardiac translational and regeneration research. Nature Protocols 11: 542-552.
Porrello E. R. (2013). microRNAs in cardiac development and regeneration. Clinical Science 125: 151-166.
Porrello E. R., Johnson B. A., Aurora A. B., Simpson E., Nam Y.J., Matkovich S. J., Dorn G. W., van Rooij E., Olson E. N. (2011a). miR-15 Family Regulates Postnatal Mitotic Arrest of Cardiomyocytes. Circulation Research 109: 670-679.
Porrello E. R., Mahmoud A. I., Simpson E., Hill J. A., Richardson J. A., Olson E. N., Sadek H. A. (2011b). Transient Regenerative Potential of the Neonatal Mouse Heart. Science 331: 1078-1080.
Porrello E. R., Mahmoud A. I., Simpson E., Johnson B. A., Grinsfelder D., Canseco D., Mammen P. P., Rothermel B. A., Olson E. N., Sadek H. A. (2013). Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proceedings of the National Academy of Sciences 110: 187-192.
Poss K. D., Wilson L. G., Keating M. T. (2002). Heart Regeneration in Zebrafish. Science 298: 2188-2190.
Przybyt E., Krenning G., Brinker M. G.L., Harmsen M. C. (2013). Adipose stromal cells primed with hypoxia and inflammation enhance cardiomyocyte proliferation rate in vitro through STAT3 and Erk1/2. Journal of Translational Medicine 11: 39.
Qian L., Huang Y., Spencer C. I., Foley A., Vedantham V., Liu L., Conway S. J., Fu J., Srivastava D. (2012). In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 485: 593-598.
Qian L., Wythe J. D., Liu J., Cartry J., Vogler G., Mohapatra B., Otway R. T., Huang Y., King I. N., Maillet M., Zheng Y., Crawley T., Taghli-Lamallem O., Semsarian C., Dunwoodie S., Winlaw D., Harvey R. P., Fatkin D., Towbin J. A., Molkentin J. D., Srivastava D., Ocorr K., Bruneau B. G., Bodmer R. (2011). Tinman/Nkx2-5 acts via miR-1 and upstream of Cdc42 to regulate heart function across species. Journal of Cell Biology 193: 1181-1196.
Rane S., He M., Sayed D., Vashistha H., Malhotra A., Sadoshima J., Vatner D. E., Vatner S. F., Abdellatif M. (2009). Downregulation of MiR-199a Derepresses Hypoxia-Inducible Factor-1α and Sirtuin 1 and Recapitulates Hypoxia Preconditioning in Cardiac Myocytes. Circulation Research 104: 879-886.
Raulf A., Horder H., Tarnawski L., Geisen C., Ottersbach A., Röll W., Jovinge S., Fleischmann B. K., Hesse M. (2015). Transgenic systems for unequivocal identification of cardiac myocyte nuclei and analysis of cardiomyocyte cell cycle status. Basic Research in Cardiology 110: 33.
Ren X.P., Wu J., Wang X., Sartor M. A., Qian J., Jones K., Nicolaou P., Pritchard T. J., Fan G.C. (2009). MicroRNA-320 Is Involved in the Regulation of Cardiac Ischemia/Reperfusion Injury by Targeting Heat-Shock Protein 20. Circulation 119: 2357-2366.
Riehle C., Wende A. R., Zhu Y., Oliveira K. J., Pereira R. O., Jaishy B. P., Bevins J., Valdez S., Noh J., Kim B. J., Moreira A. B., Weatherford E. T., Manivel R., Rawlings T. A., Rech M., White M. F., Abel E. D. (2014). Insulin Receptor Substrates Are Essential for the Bioenergetic and Hypertrophic Response of the Heart to Exercise Training. Molecular and Cellular Biology 34: 3450-3460.
Romagnuolo R., Masoudpour H., Porta-Sánchez A., Qiang B., Barry J., Laskary A., Qi X., Massé S., Magtibay K., Kawajiri H., Wu J., Valdman Sadikov T., Rothberg J., Panchalingam K. M., Titus E., Li R.K., Zandstra P. W., Wright G. A., Nanthakumar K., Ghugre N. R., Keller G., Laflamme M. A. (2019). Human Embryonic Stem Cell-Derived Cardiomyocytes Regenerate the Infarcted Pig Heart but Induce Ventricular Tachyarrhythmias. Stem Cell Reports 12: 967-981.
Rose B. A., Force T., Wang Y. (2010). Mitogen-Activated Protein Kinase Signaling in the Heart: Angels Versus Demons in a Heart-Breaking Tale. Physiological Reviews 90: 1507-1546.
Saker D.M., Walsh-Sukys M., Spector M., Zahka K.G. (1997). Cardiac Recovery and Survival After Neonatal Myocardial Infarction. Pediatric Cardiology 18: 139-142.
Schiattarella G. G., Hill J. A. (2015). Inhibition of Hypertrophy Is a Good Therapeutic Strategy in Ventricular Pressure Overload. Circulation 131: 1435-1447.
Sehring I. M., Jahn C., Weidinger G. (2016). Zebrafish fin and heart: what's special about regeneration?. Current Opinion in Genetics & Development 40: 48-56.
Sen S., Sadek H. A. (2015). Neonatal Heart Regeneration: Mounting Support and Need for Technical Standards. Journal of the American Heart Association 4: e001727.
Senyo S. E., Steinhauser M. L., Pizzimenti C. L., Yang V. K., Cai L., Wang M., Wu T.D., Guerquin-Kern J.L., Lechene C. P., Lee R. T. (2013). Mammalian heart renewal by pre-existing cardiomyocytes. Nature 493: 433-436.
Shen Y., Dong L.F., Zhou R.M., Yao J., Song Y.C., Yang H., Jiang Q., Yan B. (2016). Role of long non‐coding RNA MIAT in proliferation, apoptosis and migration of lens epithelial cells: a clinical and in vitro study . Journal of Cellular and Molecular Medicine 20: 537-548.
Sheu-Gruttadauria J., MacRae I. J. (2018). Phase Transitions in the Assembly and Function of Human miRISC. Cell 173: 946-957.e16.
Shi X. J. H., Xing W., Ding Y., Zhang W., Le X. (2007). Observation of cytodynamics in goat ventricular muscle cells. Journal of Zhengzhou University (Medical Sciences) 142: 4.
Song K., Nam Y.J., Luo X., Qi X., Tan W., Huang G. N., Acharya A., Smith C. L., Tallquist M. D., Neilson E. G., Hill J. A., Bassel-Duby R., Olson E. N. (2012). Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485: 599-604.
Souders C. A., Bowers S. L.K., Baudino T. A. (2009). Cardiac Fibroblast. Circulation Research 105: 1164-1176.
Spalding K. L., Bhardwaj R. D., Buchholz B. A., Druid H., Frisén J. (2005a). Retrospective Birth Dating of Cells in Humans. Cell 122: 133-143.
Spalding K. L., Buchholz B. A., Bergman L.E., Druid H., Frisén J. (2005b). Age written in teeth by nuclear tests. Nature 437: 333-334.
Sylva M., van den Hoff M. J.B., Moorman A. F.M. (2014). Development of the human heart. American Journal of Medical Genetics Part A 164: 1347-1371.
Tahara N., Brush M., Kawakami Y. (2016). Cell migration during heart regeneration in zebrafish. Developmental Dynamics 245: 774-787.
Takaya T., Ono K., Kawamura T., Takanabe R., Kaichi S., Morimoto T., Wada H., Kita T., Shimatsu A., Hasegawa K. (2009). MicroRNA-1 and MicroRNA-133 in Spontaneous Myocardial Differentiation of Mouse Embryonic Stem Cells. Circulation Journal 73: 1492-1497.
Takimoto E., Koitabashi N., Hsu S., Ketner E. A., Zhang M., Nagayama T., Bedja D., Gabrielson K. L., Blanton R., Siderovski D. P., Mendelsohn M. E., Kass D. A. (2009). Regulator of G protein signaling 2 mediates cardiac compensation to pressure overload and antihypertrophic effects of PDE5 inhibition in mice. Journal of Clinical Investigation 119: 408-420.
Tamirisa P., Blumer K. J., Muslin A. J. (1999). RGS4 Inhibits G-Protein Signaling in Cardiomyocytes. Circulation 99: 441-447.
Tan C. M. J., Lewandowski A. J. (2020). The Transitional Heart: From Early Embryonic and Fetal Development to Neonatal Life. Fetal Diagnosis and Therapy 47: 373-386.
Tao H., Zhang J.G., Qin R.H., Dai C., Shi P., Yang J.J., Deng Z.Y., Shi K.H. (2017). LncRNA GAS5 controls cardiac fibroblast activation and fibrosis by targeting miR-21 via PTEN/MMP-2 signaling pathway. Toxicology 386: 11-18.
Tao Z., Loo S., Su L., Abdurrachim D., Lalic J., Lee T. H., Chen X., Tan R.S., Zhang J., Ye L. (2020). Dexamethasone inhibits regeneration and causes ventricular aneurysm in the neonatal porcine heart after myocardial infarction. Journal of Molecular and Cellular Cardiology 144: 15-23.
Terzic A., Behfar A. (2016). Stem cell therapy for heart failure: Ensuring regenerative proficiency. Trends in Cardiovascular Medicine 26: 395-404.
Tian Y., Liu Y., Wang T., Zhou N., Kong J., Chen L., Snitow M., Morley M., Li D., Petrenko N., Zhou S., Lu M., Gao E., Koch W. J., Stewart K. M., Morrisey E. E. (2015). A microRNA-Hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Science Translational Medicine 7: 279ra238.
Ulrich V., Rotllan N., Araldi E., Luciano A., Skroblin P., Abonnenc M., Perrotta P., Yin X., Bauer A., Leslie K. L., Zhang P., Aryal B., Montgomery R. L., Thum T., Martin K., Suarez Y., Mayr M., Fernandez‐Hernando C., Sessa W. C. (2016). Chronic miR‐29 antagonism promotes favorable plaque remodeling in atherosclerotic mice. EMBO Molecular Medicine 8: 643-653.
Uygur A., Lee R. T. (2016). Mechanisms of Cardiac Regeneration. Developmental Cell 36: 362-374.
van Rooij E., Sutherland L. B., Qi X., Richardson J. A., Hill J., Olson E. N. (2007). Control of Stress-Dependent Cardiac Growth and Gene Expression by a MicroRNA. Science 316: 575-579.
Velayutham N., Agnew E. J., Yutzey K. E. (2019). Postnatal Cardiac Development and Regenerative Potential in Large Mammals. Pediatric Cardiology 40: 1345-1358.
Velayutham N., Alfieri C. M., Agnew E. J., Riggs K. W., Baker R. S., Ponny S. R., Zafar F., Yutzey K. E. (2020). Cardiomyocyte cell cycling, maturation, and growth by multinucleation in postnatal swine. Journal of Molecular and Cellular Cardiology 146: 95-108.
Ventura A., Young A. G., Winslow M. M., Lintault L., Meissner A., Erkeland S. J., Newman J., Bronson R. T., Crowley D., Stone J. R., Jaenisch R., Sharp P. A., Jacks T. (2008). Targeted Deletion Reveals Essential and Overlapping Functions of the miR-17∼92 Family of miRNA Clusters. Cell 132: 875-886.
Vitali V., Rothering R., Catania F. (2021). Fifty Generations of Amitosis: Tracing Asymmetric Allele Segregation in Polyploid Cells with Single-Cell DNA Sequencing. Microorganisms 9: 1979.
Vliegen H. W., Eulderink F., Bruschke A. V., Van der, Laarse A., Cornelisse C. J. (1995). Polyploidy of myocyte nuclei in pressure overloaded human hearts: a flow cytometric study in left and right ventricular myocardium. The American journal of cardiovascular pathology 5: 27-31.
Wang F., Liu S., Pei J., Cai L., Liu N., Liang T., Dong X., Cong X., Chun J., Chen J., Hu S., Chen X. (2020a). LPA 3 -mediated lysophosphatidic acid signaling promotes postnatal heart regeneration in mice . Theranostics 10: 10892-10907.
Wang J., Greene S. B., Bonilla-Claudio M., Tao Y., Zhang J., Bai Y., Huang Z., Black B. L., Wang F., Martin J. F. (2010). Bmp Signaling Regulates Myocardial Differentiation from Cardiac Progenitors Through a MicroRNA-Mediated Mechanism. Developmental Cell 19: 903-912.
Wang J.X., Zhang X.J., Li Q., Wang K., Wang Y., Jiao J.Q., Feng C., Teng S., Zhou L.Y., Gong Y., Zhou Z.X., Liu J., Wang J.L., Li P. (2015a). MicroRNA-103/107 Regulate Programmed Necrosis and Myocardial Ischemia/Reperfusion Injury Through Targeting FADD. Circulation Research 117: 352-363.
Wang K., Liu C.Y., Zhou L.Y., Wang J.X., Wang M., Zhao B., Zhao W.K., Xu S.J., Fan L.H., Zhang X.J., Feng C., Wang C.Q., Zhao Y.F., Li P.F. (2015b). APF lncRNA regulates autophagy and myocardial infarction by targeting miR-188-3p. Nature Communications 6: 7779.
Wang K., Liu F., Zhou L.Y., Long B., Yuan S.M., Wang Y., Liu C.Y., Sun T., Zhang X.J., Li P.F. (2014a). The Long Noncoding RNA CHRF Regulates Cardiac Hypertrophy by Targeting miR-489. Circulation Research 114: 1377-1388.
Wang K., Long B., Zhou L.Y., Liu F., Zhou Q.Y., Liu C.Y., Fan Y.Y., Li P.F. (2014b). CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nature Communications 5: 3596.
Wang S., Talukder A., Cha M., Li X., Hu H. (2021). Computational annotation of miRNA transcription start sites. Briefings in Bioinformatics 22: 380-392.
Wang Y., Li Y., Feng J., Liu W., Li Y., Liu J., Yin Q., Lian H., Liu L., Nie Y. (2020b). Mydgf promotes Cardiomyocyte proliferation and Neonatal Heart regeneration. Theranostics 10: 9100-9112.
Weber K., Sun Y., Katwa L., Cleutjens J. (1995). Connective tissue: a metabolic Entity?. Journal of Molecular and Cellular Cardiology 27: 107-120.
Wei K., Serpooshan V., Hurtado C., Diez-Cuñado M., Zhao M., Maruyama S., Zhu W., Fajardo G., Noseda M., Nakamura K., Tian X., Liu Q., Wang A., Matsuura Y., Bushway P., Cai W., Savchenko A., Mahmoudi M., Schneider M. D., van den Hoff M. J. B., Butte M. J., Yang P. C., Walsh K., Zhou B., Bernstein D., Mercola M., Ruiz-Lozano P. (2015). Epicardial FSTL1 reconstitution regenerates the adult mammalian heart. Nature 525: 479-485.
Whelan R. S., Kaplinskiy V., Kitsis R. N. (2010). Cell Death in the Pathogenesis of Heart Disease: Mechanisms and Significance. Annual Review of Physiology 72: 19-44.
Wilkins B. J., Dai Y.S., Bueno O. F., Parsons S. A., Xu J., Plank D. M., Jones F., Kimball T. R., Molkentin J. D. (2004). Calcineurin/NFAT Coupling Participates in Pathological, but not Physiological, Cardiac Hypertrophy. Circulation Research 94: 110-118.
Wu Y., Zhou L., Liu H., Duan R., Zhou H., Zhang F., He X., Lu D., Xiong K., Xiong M., Zhuang J., Liu Y., Li L., Liang D., Chen Y.H. (2021). LRP6 downregulation promotes cardiomyocyte proliferation and heart regeneration. Cell Research 31: 450-462.
Xu C., Lu Y., Pan Z., Chu W., Luo X., Lin H., Xiao J., Shan H., Wang Z., Yang B. (2007). The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes . Journal of Cell Science 120: 3045-3052.
Ye L., D’Agostino G., Loo S. J., Wang C. X., Su L. P., Tan S. H., Tee G. Z., Pua C. J., Pena E. M., Cheng R. B., Chen W. C., Abdurrachim D., Lalic J., Tan R. S., Lee T. H., Zhang J.Y., Cook S. A. (2018). Early Regenerative Capacity in the Porcine Heart. Circulation 138: 2798-2808.
Yin V. P., Lepilina A., Smith A., Poss K. D. (2012). Regulation of zebrafish heart regeneration by miR-133. Developmental Biology 365: 319-327.
Zacchigna S., Martinelli V., Moimas S., Colliva A., Anzini M., Nordio A., Costa A., Rehman M., Vodret S., Pierro C., Colussi G., Zentilin L., Gutierrez M. I., Dirkx E., Long C., Sinagra G., Klatzmann D., Giacca M. (2018). Paracrine effect of regulatory T cells promotes cardiomyocyte proliferation during pregnancy and after myocardial infarction. Nature Communications 9: 2432.
Zacchigna S., Zentilin L., Giacca M. (2014). Adeno-Associated Virus Vectors as Therapeutic and Investigational Tools in the Cardiovascular System. Circulation Research 114: 1827-1846.
Zeigler A. C., Richardson W. J., Holmes J. W., Saucerman J. J. (2016). Computational modeling of cardiac fibroblasts and fibrosis. Journal of Molecular and Cellular Cardiology 93: 73-83.
Zhao Y., Ransom J. F., Li A., Vedantham V., von Drehle M., Muth A. N., Tsuchihashi T., McManus M. T., Schwartz R. J., Srivastava D. (2007). Dysregulation of Cardiogenesis, Cardiac Conduction, and Cell Cycle in Mice Lacking miRNA-1-2. Cell 129: 303-317.
Zhao Y., Samal E., Srivastava D. (2005). Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 436: 214-220.
Zhao Y., Sawyer D. R., Baliga R. R., Opel D. J., Han X., Marchionni M. A., Kelly R. A. (1998). Neuregulins Promote Survival and Growth of Cardiac Myocytes. Journal of Biological Chemistry 273: 10261-10269.
Zhu W., Zhang E., Zhao M., Chong Z., Fan C., Tang Y., Hunter J. D., Borovjagin A. V., Walcott G. P., Chen J. Y., Qin G., Zhang J. (2018). Regenerative Potential of Neonatal Porcine Hearts. Circulation 138: 2809-2816.