Int. J. Dev. Biol. 66: 59 - 75 (2022)
Special Issue: Developmental Biology in Greece
Developmental and regenerative biology of cardiomyocytes
Review | Published: 26 October 2021
Abstract
Current progress and challenges in understanding the molecular and cellular mechanisms of cardiomyocyte embryonic development and regeneration are reviewed in our present work. Three major topics are critically discussed: how do cardiomyocytes form in the embryo? What is the adult origin of the cells that regenerate cardiomyocytes in animal models with adult heart regeneration capabilities? Can the promise of therapeutic cardiomyocyte regeneration be realized in humans? In the first topic, we highlight current advances in understanding the developmental biology of cardiomyocytes, with emphasis on the regulative capabilities of the early embryo during specification and allocation of the cardiomyoblasts that produce the primordial heart. We place further emphasis on trabecular cardiomyocyte development from late cardiomyoblasts, neural crest cells and primordial cardiomyocytes, and their critical role in the clonal growth of the compact/septal and cortical cardiomyocyte layers in the mammalian embryo and adult zebrafish, respectively. In the second topic, we focus on the re-activation of the cortical or trabecular compaction programs as hallmarks of cardiomyocyte regenerative cells during adult zebrafish and neonatal mouse heart regeneration, respectively, and underscore the metabolic remodeling that commonly drives cardiomyocyte regeneration in these organisms. Finally, we discuss the status of preclinical and clinical-stage therapeutics for cardiomyocyte regeneration, with particular emphasis on gene therapy, as well as adult and pluripotent stem cell-based cellular cardiomyoplasty approaches. In summary, our article provides a bird’s-eye view of current knowledge and potential pitfalls in the field of developmental biology-guided regenerative medicine strategies for the treatment of heart diseases.
Keywords
Heart development, cardiomyocyte regeneration, cardiac regenerative medicine, cell therapy, gene therapy
Introduction
The lack of cardiomyocyte (CM) regenerative capabilities in the adult heart is currently considered to be the most vulnerable aspect of human life (Eschenhagen et al. 2017). CMs comprise the building blocks of the heart muscle and, once formed in the early embryo, are tasked with the lifelong responsibility of remaining healthy and contractile to keep the heart pumping blood throughout the body (Hashimoto et al. 2018). Unlike most other adult tissues, humans generate limited amounts of heart muscle after birth, with recent estimates suggesting that more than half of the CMs in a ~70-year-old healthy human are remnants of embryogenesis (Eschenhagen et al. 2017). Hence, any loss of CMs after birth due to disease or injury is irreparable and, depending on its extent, may be lethal or be replaced with dysfunctional scar tissue that will eventually lead to heart failure (HF) (Hashimoto et al. 2018).
Accordingly, there is a great need to address the ever-growing heart disease epidemic and develop novel cardiac regenerative medicine approaches that could revive the failing heart (Hashimoto et al. 2018). To this end, animal embryos and human pluripotent stem cells (hPSCs); as well as animal models with full postnatal cardiac regenerative capabilities such as axolotls, zebrafish, and 1-day old mouse pups and pigs; are at the forefront of research to unravel the relationship between cell sources that produce new CMs in embryos and adults (Sadek and Porrello, 2020). Consequently, basic research findings are guiding the development of novel therapeutic strategies for CM regeneration, including the manufacture of theoretically unlimited amounts of lab-grown hPSC-derived CMs as grafts for direct scar remuscularization (Yamanaka, 2020); as well as pharmaceutical and gene therapies to stimulate endogenous mechanisms of cardiomyogenesis in the adult heart (Hashimoto et al. 2018).
Here, we provide a comprehensive review of current progress and challenges in the field of cardiac regenerative medicine. We discuss new mechanistic insights in the development and regeneration of CMs in zebrafish and neonatal mice, and provide our critical view of potential pharmaceutical, cell- and gene-based cardiac regenerative medicine strategies that are currently in preclinical or clinical stages of experimental testing.
Early development of cardiomyocytes
In most animals in which cell lineages have been fate-mapped, the cells that give rise to CMs appear to be specified early during development, before the beginning of gastrulation. For example, sea squirts (Ciona intestinalis or C.intestinalis) present an ideal system for the study of developmental heart genetics, due to their unique evolutionary position as invertebrate chordates. Consequently, they can be viewed as a minimalistic version of vertebrates, before the evolution of large-scale gene duplications that introduced redundancies and additional levels of complexity in vertebrate gene function (Christiaen et al. 2009). In these species, maternal mRNAs for β-catenin and Macho1, which are highly enriched in the yellow egg cytoplasm, are passed down during cleavage to the B7.5 pair of heart muscle-producing blastomeres to directly activate their target genes, the LIM-homeobox Lhx3 and the T-box transcription factor Tbx6c, respectively. Lhx3 and Tbx6c act synergistically to activate the basic helix-loop-helix (bHLH) transcription factor mesoderm posterior (Mesp), specifying the B7.5 blastomeres as the earliest progenitors of the myocardial lineage. Upon gastrulation, the B7.5 blastomeres divide asymmetrically and form a pair of Mesp-expressing progeny on each side of the embryo. Their rostral descendants will receive Fgf/Ets signals from the endoderm and migrate anteriorly to form the bilateral heart fields, while caudal descendants produce the pharyngeal muscles. Mesp and FGF/Ets will synergistically activate a cardiac-specific enhancer of the forkhead box-F (FoxF) transcription factor, and FoxF will then instruct the reconstitution of the C.intestinalis cardiac gene regulatory network (GRN). Remarkably, the C.intestinalis cardiac GRN closely matches closely that of vertebrates and includes the master cardiac transcription factors myocardin, Gata, Nkx2.5, the bHLH factors Hand1 and Hand2, and the Mef2 genes. The two heart fields proliferate to ~12-32 cells and will eventually fuse in the midline to form a linear heart tube that becomes C.intestinalis’s rudimentary heart, built as a single layer of contractile cardiac myoepithelial cells under an epithelium of pericardial cells (Christiaen et al. 2009).
A similar sequence of events underlies vertebrate heart development. In chick embryos, lineage-tracing and cell transplantation experiments have pinpointed the prospective heart cells as the lateral anterior cells in the primitive streak (PS) stage embryo (Ehrman and Yutzey, 1999). In mice, myocardial progenitors are specified several hours before PS formation in a subset of proximal posterior epiblast cells, which activate the T-box transcription factor Eomesodermin (Eomes) in response to Activin/Nodal and Wnt/β-catenin signaling. Subsequently, Eomes directly binds to T-box consensus binding sites present within an evolutionary conserved early mesoderm enhancer of the Mesp1 gene (Costello et al. 2011). Mesp1 acts to prevent endoderm specification of the prospective lateral plate mesoderm cells by directly binding and repressing expression of the genes Brachyury, Sox17, and Goosecoid, while activating the canonical Wnt signaling inhibitor Dickkopf-related protein 1 (Dkk1), to inhibit hemangioblastic specification (David et al. 2008). Compared to C.intestinalis, mice have a single Tbx6 gene and 2 Mesp paralogs, Mesp1 and Mesp2. Tbx6 binds and directly activates the Mesp2 gene, which drives paraxial mesoderm specification (Costello et al. 2011). Upon gastrulation, ~250 Mesp1-expressing cells migrate in two spatiotemporally distinct pools. The first pool migrates on embryonic day (E) 6.5, as part of the lateral plate splanchnic mesoderm lineage and forms a pair of Mesp1-expressing heart fields on each side of the embryo, before fusing in the midline to form the cardiac crescent or first heart field (FHF), which will eventually give rise to a linear heart tube. The second pool migrates at ~E7.5 dorsally and medially to the FHF, as part of the pharyngeal mesoderm -which is part of the head mesoderm and comprises a mixture of lateral plate and paraxial mesoderm lineages- to form a secondary heart field (SHF) (Chabab et al. 2016). FHF progenitors upregulate expression of the T-box transcription factor Tbx5 and produce the left ventricular as well as some of the right and left atrial CMs. Meanwhile, the SHF activates regulatory sequences for Tbx1 and the LIM homeodomain transcription factor Isl1 and produces the right ventricular and the remainder of the atrial CMs. In zebrafish, the FHF generates most of the single ventricular CMs, whereas most of the atrial CMs and the distal-most part of the ventricle, which connects to the atria, come from the SHF (Sanchez-Iranzo et al. 2018). Lineage-tracing studies in mice and in vitro pluripotent stem cell-based modeling experiments of human cardiomyogenesis indicate that Mesp1 descendants positioned more rostrally to the FHF and SHF activate the transcription factor Foxa2 that controls the ventricular CMs GRN. On the other hand, those that end up more caudally encounter a retinoic acid (RA) gradient and activate the atrial CMs GRN (Bardot et al. 2017; Lee et al. 2017).
The SHF is patterned into anterior (aSHF) and posterior (pSHF) domains along the arterial and venous poles of the FHF-derived linear heart tube, respectively (Hatzistergos et al. 2020). Both domains are kept in a proliferative progenitor cell state for several days as they become the main resource for the new CMs needed for the elongation of the heart tube and subsequent development of the right ventricle (RV) and atria. In the aSHF, this is achieved partly through synergism between Wnt/β-catenin and Fgf10 signaling, which are maintained active by Isl1 and Tbx1, whereas the pSHF is maintained through Wnt/β-catenin and RA-mediated activation of Hox genes (Bertrand et al. 2011). Notably, in aSHF and pSHF cardioblasts Isl1 is activated through distinct cis-regulatory elements (Hatzistergos et al. 2020). In addition, the aSHF is regulated through oxygen tension (Yuan et al. 2017). Within the aSHF domain, O2 concentration is relatively high, leading to continuous degradation of the hypoxia inducible transcription factor-1α (Hif-1α). The absence of Hif-1α allows Isl1 to remain active and recruit the histone deacetylases Hdac1/Hdac5 to promoter sequences for the master cardiac transcription factor Nkx2.5, thereby preventing its expression. Upon migration into the heart tube, the levels of O2 decrease, leading to stabilization of Hif-1α. Hif-1α translocates to the nucleus and interacts with the Notch effector Hes family bHLH transcription factor 1 (Hes1) and the protein deacetylase sirtuin 1 (Sirt1) to repress Isl1 expression. In parallel, Hif-1α directly binds to hypoxia response elements within the promoter elements of the Nkx2.5 gene and recruits the histone acetyltransferase (HAT) p300, activating its expression (Yuan et al. 2017). Moreover, Nkx2.5 directly activates a small, atypical homeodomain protein without homeobox DNA binding capacity, called Hopx (Jain et al. 2015). As aSHF progenitors migrate into the heart tube, they encounter a Bmp2/4 signaling zone that activates Smad1/5/8 and co-Smad4 in their cytoplasm. Hopx co-operates with Smad4 and Hdac2 and, together, they move into the nucleus to bind and suppress the expression of several canonical Wnt signaling genes. This causes aSHF progenitors to stop dividing and undergo specification into CMs. Concurrently, to avoid potential exhaustion of the aSHF pool from excessive Hopx-Smad-Hdac signaling, Nkx2.5 establishes a negative feedback loop to directly suppress Bmp2/Smad signaling, thereby enabling cardioblasts to proliferate while undergoing specification into CMs (Prall et al. 2007; Jain et al. 2015).
Studies in fish and salamanders have demonstrated that the development of heart fields is regulative (Ehrman and Yutzey, 1999; Sanchez-Iranzo et al. 2018). Genetic ablation or extirpation of one or both heart fields during early development results in normal cardiogenesis, indicating that FHF progenitors can be fully compensated by SHF progenitors and vice versa. Similarly, extirpation of the prospective cardiogenic mesoderm can be fully compensated through the recruitment of mesodermal cells from adjacent regions that normally do not contribute to cardiogenesis (Ehrman and Yutzey, 1999; Sanchez-Iranzo et al. 2018).
Notably, studies in chicken embryos show that the different types of mesoderm are specified by opposing Bmp and Noggin gradients, which produce an increasing Bmp concentration along the mediolateral axis, with the highest levels of Bmp activity reaching the lateral-most extraembryonic mesoderm layer, followed by lateral plate, intermediate and paraxial mesoderm layers, respectively (Tonegawa and Takahashi, 1998) (Fig. 1). Fate-mapping experiments, combined with in situ hybridization detection of Bmp2 and Nkx2.5 transcripts, indicate that the medial border of extraembryonic tissue demarcates the lateral-most border of the heart-forming region, whereas the medial FHF border is placed at the lateral edge of the mesoderm overlying the prospective neural plate. Complete or partial removal of this Bmp2/Nkx2.5-expressing FHF domain results in loss of corresponding cardiac structures, consistent with the notion that development of the FHF in birds is not regulative (Ehrman and Yutzey, 1999). Intriguingly, however, recent genetic fate-mapping and single-cell transcriptomic studies in mouse embryos show that cells from the extraembryonic mesoderm adjacent to the lateral-most border of the heart forming region contribute to the FHF, suggesting that at least some level of regulative capacity is present in the mammalian cardiogenic mesoderm. These regulatory cells are marked by the expression of the bHLH transcription factor Hand1 (Fig. 1), an essential component of the cardiac GRN (Tyser et al. 2021; Zhang et al. 2021). Consistent with these findings, global knockout of Hand1 leads to embryonic lethality at approximately E8.5 due to deficiencies in extraembryonic and cardiogenic mesoderm layers (Firulli et al. 1998).
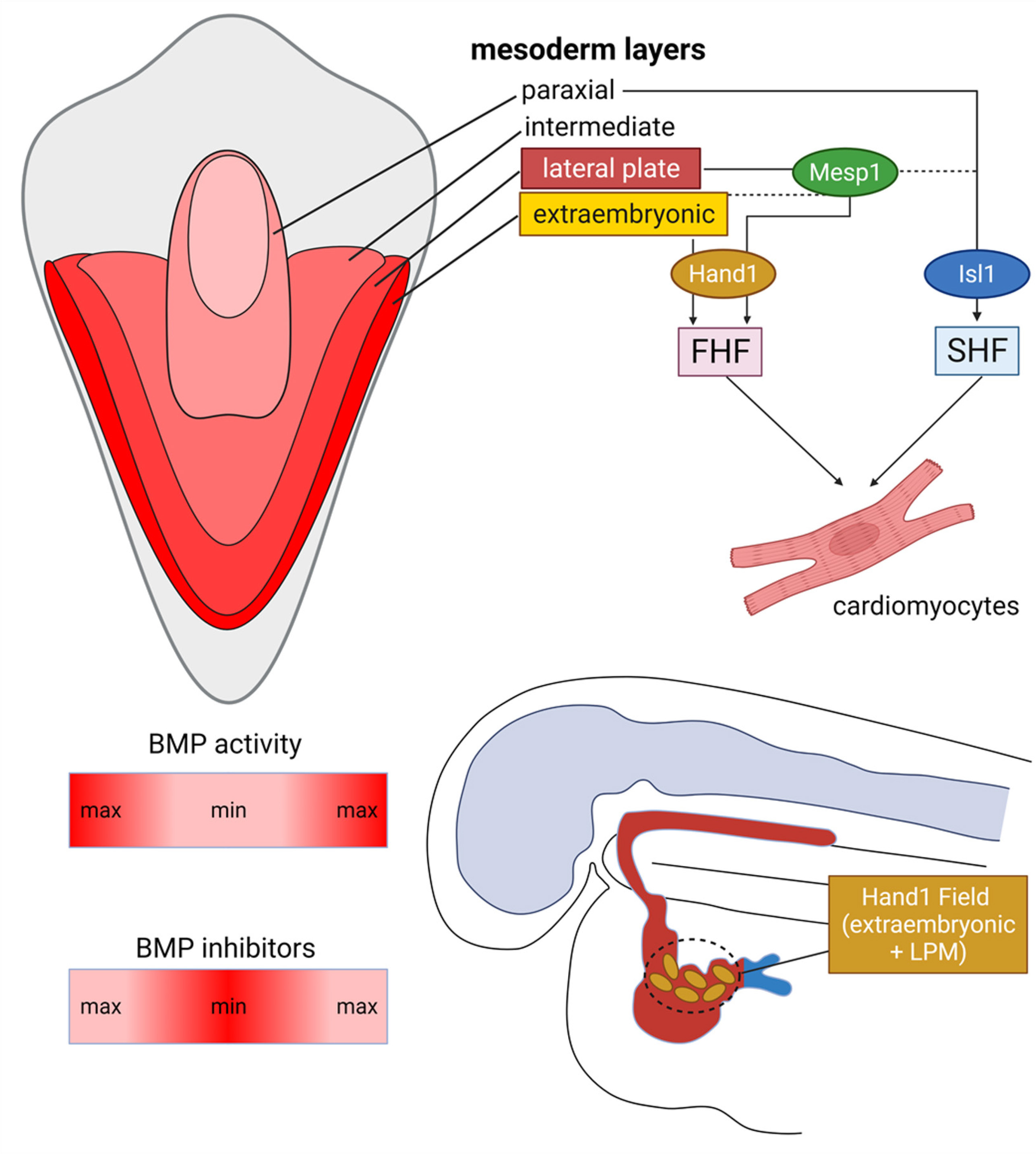
Fig. 1. Early development of cardiomyocytes in mammals.
A dorsoventral Bmp gradient specifies different mesoderm subtypes in the gastrulating embryo. First Heart Field (FHF) progenitors are specified within the anterior lateral plate (LPM) but also recruit cells from a Hand1-expressing domain established more ventrally, at the LPM-extraembryonic mesoderm junction. First Heart Field Hand1 transcription factor generates dorsal and lateral CMs of the primordial heart tube. Similarly, secondary heart field (SHF) progenitors are specified within the cardiopharyngeal mesoderm, an Isl1-expressing domain established dorsomedially to the FHF, at the junction of LPM and paraxial mesoderm.
Growth of new cardiomyocytes during cardiac chamber morphogenesis
Genetic lineage-tracing experiments in mice reveal that embryonic development of mammalian CMs from FHF and SHF cardiac progenitors peaks at ~E8.0, when cycling RV cardiomyoblasts from the SHF are incorporated into the heart tube, and gradually declines thereafter before termination at ~E11.5 (Li et al. 2019; Hatzistergos et al. 2020). At that stage, the elongated heart tube bends rightward forming an s-shaped loop. The looped heart enters its growth and chamber morphogenesis phase, so that the rapidly increasing metabolic demands of the growing embryo are met by a proportional increase in oxygen and nutrients through blood supply. In fish and amphibians, this is achieved mainly through the growth of a new layer of CMs under the endocardial surface of their only ventricle. This new layer undergoes patterning into irregular ridges called trabeculae carneae, converting the inner surface of the ventricle from a smooth to a spongy structure (Fukuda et al. 2019). Compared to the smooth surface, the spongy pattern of the trabecular myocardium is thought to be ergonomically and mechanically advantageous in terms of hemodynamics, as it enables the heart to achieve greater end-diastolic and shorter end-systolic volumes, thereby enhancing cardiac output without increasing the surface area. In reptiles, birds, and mammals, trabeculation is accompanied by proliferative growth of the overlying compact myocardium and is followed by a compaction phase. During the latter, the trabecular ridges become progressively compacted into the ventricular walls following a base-to-apex direction, with those closer to the apex eventually coalescing into a single, thicker ridge that will become the muscular layer of the interventricular septum (Del Monte-Nieto et al. 2018). Moreover, trabecular CMs are clonally related to parts of the cardiac conduction system, including the Purkinje fibers. Thus, compared to anamniotes, amniote embryos adapt to their greater hemodynamic and metabolic developmental needs by deploying a range of solutions -growth of additional CMs, trabeculation, compaction, and ventricular septation- which, collectively, lead to substantial thickening of their primordial ventricular walls and complete separation (or partial in some reptiles) of the oxygenated and deoxygenated blood flows (Del Monte-Nieto et al. 2018). Inhibiting trabeculation leads to embryonic demise, while excessive trabeculation results in left ventricular noncompaction cardiomyopathy and subsequent HF (Pashmforoush et al. 2004).
More recently, clonal analysis under a Brainbow-Cre-reporter construct demonstrated that zebrafish are also producing more CMs to thicken their extensively trabeculated, single-layered primordial myocardial walls, but this occurs during juvenile growth rather than embryonically, and is not followed by trabecular compaction. Specifically, at ~6 weeks of age, adult zebrafish develop about eight CMs at the base of their ventricle, which undergo massive clonal expansion, and by ~12 weeks have enveloped the entire ventricular subepicardial surface (Gupta and Poss, 2012). Surprisingly, these rare, adult cardiomyoblasts, which will produce the entire new “cortical” layer, are clonally related to trabecular CMs. Gupta and Poss (2012) suggested that the trabecular CMs dedifferentiate, breach the primordial compact layer, and colonize the subepicardium where, in response to epicardium- derived RA signals, activate regulatory sequences for the Gata4 cardiac transcription factor before expanding clonally (Gupta and Poss, 2012). Notably, as will be discussed in the following sections, the mechanisms by which adult zebrafish trabecular CMs become plastic and generate Gata4+ proliferative cortical cardiomyoblasts are also evoked in response to stress or injury, and this capability is thought to underlie the full regenerative potential of the adult zebrafish heart (Gupta and Poss, 2012).
In zebrafish, the developmental origins of these rare, cortical layer-producing adult trabecular CMs are likely heterogeneous with some descending from the neural crest (NC) (Sande-Melon et al. 2019) and others from the Tbx5a+ FHF lineages (Sanchez-Iranzo et al. 2018). The NC comprises a population of vertebrate-specific migratory stem cells, which develop transiently during neurulation, from the neural ectoderm cells which end up at the interface of the closing neural tube and surface ectoderm and undergo epithelial-to-mesenchymal transition (EMT) before migrating into most developing organs, including the heart (Hatzistergos et al. 2020). Interestingly, although the Tbx5a+ FHF-derived cortical lineage is dispensable, since their genetic ablation in early embryos can be fully compensated by SHF-derived cells (Sanchez-Iranzo et al. 2018), the contribution of NC in trabecular CMs is indispensable. Fish, in which embryonic NC-derived CMs have been genetically ablated, develop normally but exhibit aberrant trabeculation patterns and severe hypertrophic cardiomyopathy. More importantly, loss of NC-CMs results in loss of cardiac regenerative potential in adults (Abdul-Wajid et al. 2018; Sande-Melon et al. 2019).
A similar role for the mammalian NC has also been shown in mice, in which a small number of Wnt1-Cre-tracked cells migrate into the inflow region of the developing heart (Fig. 2) and upregulate regulatory sequences for Isl1, before contributing a limited number of highly proliferative trabecular CMs (Hatzistergos et al. 2020). However, compared to fish, clonal expansion of mouse NC-derived trabecular CMs occurs embryonically rather than after birth, and is essential for building the muscular layer of the interventricular septum. Moreover, instead of breaching through the compact layer and undergoing epiboly-like, basicoapical cortical expansion, mammalian NC-derived trabecular CMs burst into large, wedge-like transmural clones with a narrow luminal and wider cortical surface (Hatzistergos et al. 2020). Intriguingly, single-cell transcriptome-based lineage-trajectory reconstruction studies suggest that a small pool of Isl1-expressing NC derivatives linger in the postnatal mouse inflow region through a positive feedback loop between Isl1 and Wnt/β-catenin (Hatzistergos et al. 2020). More importantly, their genetic fate-map through Isl1-MerCreMer reporter alleles showed that they remain plastic, as they make rare contributions in ventricular CMs. However, unlike fish and mouse embryos, CMs generated from mouse NC derivatives after birth do not exhibit clonal expansion capacity (Hatzistergos et al. 2020). Further studies will determine whether their developmental clonal expansion potential can be evoked and, like fish, contribute CMs for regeneration.
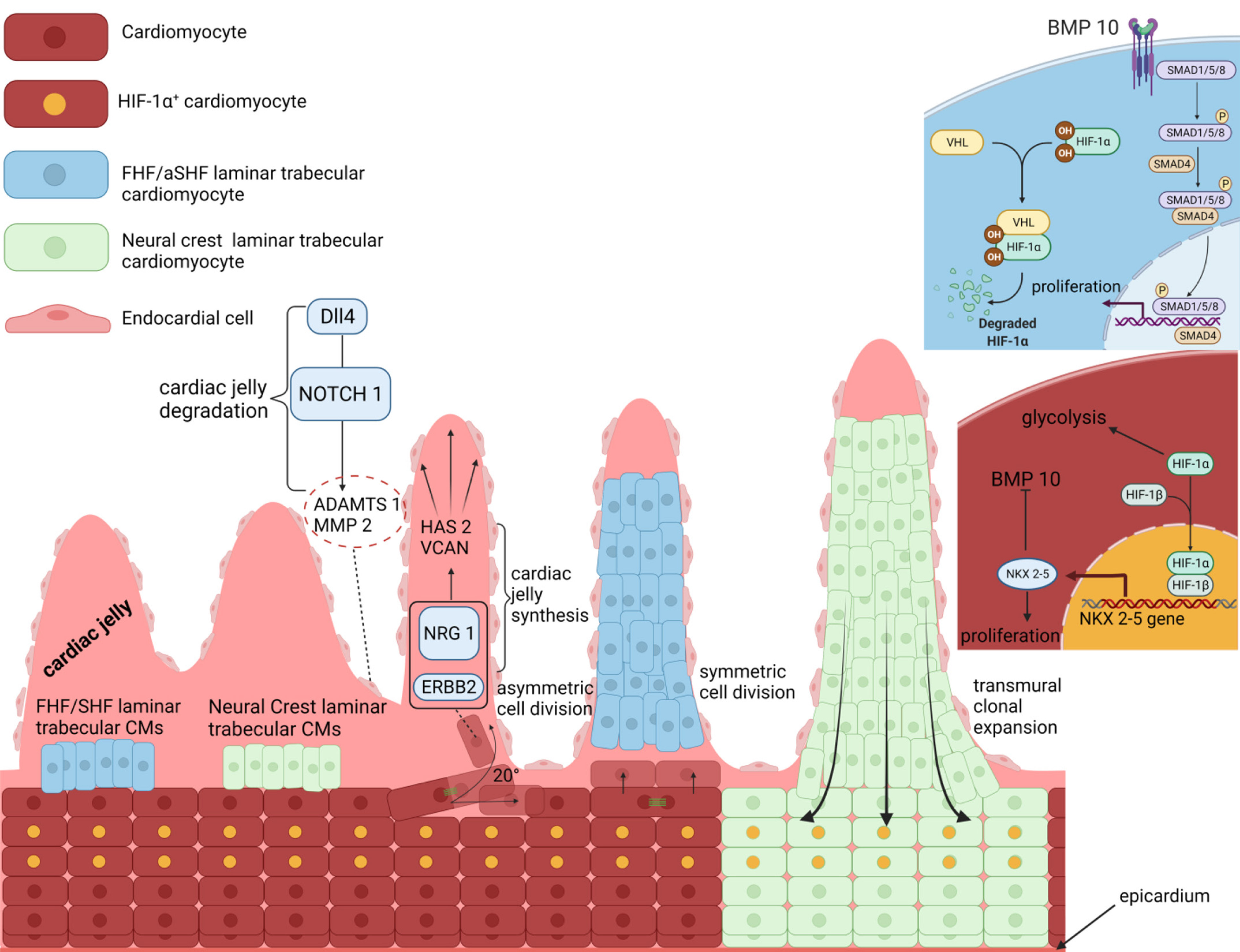
Fig. 2. Growth of trabecular and compact cardiomyocytes in mammals.
Early, laminar trabecular cardiomyocytes (CMs) develop from a small number of heart-field and neural crest (NC) cardiomyoblasts. Nrg1/ErbB2 and Dll4/Notch1 signals are activated reciprocally in CMs and endocardial cells, respectively, and act antagonistically to pattern the cardiac jelly into trabecular-CM-enclosing cysts. A Hif-1α-mediated metabolic compartmentalization controls the allocation and clonal growth of the trabecular and compact CMs. Activation of Bmp10 promotes trabecular CM proliferation, whereas inhibition of Bmp10 through the Hif-1α-mediated activation of Nkx2.5 promotes compact CM proliferation. Interestingly, trabecular and compact CMs exhibit regulative capacity, as compact CMs produce trabecular CMs through oriented cell division and/or directed migration, while trabecular CMs expand transmurally into the compact zone. FHF, first heart field; SHF, second heart field.
Although the molecular mechanisms that drive growth of trabecular CMs and ventricular chamber morphogenesis are not yet fully understood, most studies to date suggest they are controlled through a crosstalk between oxygen and surface tensions. Specifically, in both amniotes and anamniotes, trabeculation takes place exclusively in the ventricular chambers and always begins at the site of the ventricle facing the atrioventricular canal, which is the area receiving the biggest thrust from the blood flow (Del Monte-Nieto et al. 2018; Priya et al. 2020). In lower vertebrates, the current model suggests that local increases in blood flow and CM contractility cause a gradual reduction in thickness of the cardiac jelly and, at the same time, upregulation of Neuregulin (Nrg)-1/2a in endocardial cells (Priya et al. 2020). The cardiac jelly is a layer of extracellular material (ECM) that serves as a physical barrier between the primordial myocardium and the endocardial cells. Hence, its thinning reduces its ability to prevent endocardially-derived mechanical forces and signaling molecules such as Nrg1/2a from breaching into the myocardial layer, and vice versa. Consequently, increased Nrg1 concentration on the luminal surface of the ErbB2-expressing myocardial cells induces a shift in their metabolism toward glycolysis. Together, the metabolic switch and the increased mechanical tension force the prospective trabecular CMs to remodel their cytoskeleton, whereby the actomyosin filaments undergo apical constriction and their Golgi complexes are moved closer to the lateral cell membranes through the microtubules (Icardo and Fernandez-Teran, 1987; Priya et al. 2020). This activates an EMT-like process, which induces CMs to delaminate from the compact myocardial wall and seed the trabecular layer. At the same time, large intercellular spaces filled with ECM appear in the compact myocardium. The ECM is thought to be produced by the compact CMs themselves through their repositioned Golgi (Icardo and Fernandez-Teran, 1987). Moreover, an increase in Nrg2a/ErbB2 signaling triggers a marked increase in compact CM mitosis. The combination of increased CM proliferation and ECM deposition cause tissue-scale crowding and, consequently, changes in surface tension and CM contractility. Cardiomyocytes with the highest contractility and surface tension will activate the Notch lateral inhibition pathway, delaminate, and seed the trabecular layer; Notch signals, acting upon neighboring CMs, inhibit actomyosin cytoskeleton remodeling and thereby trabecular myogenesis (Icardo and Fernandez-Teran, 1987; Priya et al. 2020).
On the other hand, a substantially different model has been proposed to drive cardiac trabeculation in mammals. First and foremost, half of the trabecular CMs in mice are thought to differentiate directly from trabecular progenitor cells, while the other half extrude from compact CMs (Yue et al. 2020). In the first case, most of the trabecular progenitor cells seem to segregate early from FHF and aSHF cardiac progenitors. The first signs of trabecular CM differentiations are already visible by E8.0 in the form of “laminar trabeculae”, which are discontinuous luminal CM clusters loosely attached to the compact layer of the growing heart tube (Fig. 2) (Del Monte-Nieto et al. 2018; Yue et al. 2020). In addition, genetic fate-mapping studies from our laboratory and others indicate that NC cells will also emigrate at later stages to contribute directly to the trabecular progenitor cell pool (Fig. 2) (Abdul-Wajid et al. 2018; Tang et al. 2019; Hatzistergos et al. 2020). Intriguingly, NC cells have also been found to contribute trabecular CMs in zebrafish (Abdul-Wajid et al. 2018; Sande-Melon et al. 2019; Tang et al. 2019). Further studies at earlier stages will determine whether the current model of extrusion-based zebrafish CM trabeculation affords any improvements (Priya et al. 2020).
Compared to zebrafish, trabecular CM extrusion may occur in two different ways in the mammalian heart. About two thirds of CM extrusions occur through asymmetrically dividing compact CMs, in which the mitotic spindle of the parent cell is oriented at ~20° relative to the centroid of the left ventricle geometry, possibly in response to increased surface tension and consequent changes in their cytoskeleton. The daughter cell aiming towards the ventricular lumen becomes the trabecular progeny, while the other is retained in the compact layer (Fig. 2) (Yue et al. 2020). In addition, ~1/3 of CM extrusions occur through symmetrically dividing compact myocytes, in which both daughter cells eventually delaminate and undergo directional migration toward the cardiac lumen, resulting in trabecular formation and regional specification (Yue et al. 2020).
The signaling pathways involved also seem to operate in substantially diverse ways. For example, rather than acting as inducers and repressors of CM delamination, Nrg1/ErbB2 and Notch signals are activated reciprocally in CMs and endocardial cells, respectively, to control trabecular CM allocation by acting antagonistically on the regulation of cardiac jelly synthesis and degradation (Fig. 2) (Del Monte-Nieto et al. 2018). Specifically, Nrg1/ErbB2 signaling stimulates CMs to synthesize the cardiac jelly genes Hyaluronan synthase 2 (Has2), Versican (Vcan), and the CD44 hyaluronan receptor, while suppressing synthesis of metalloproteinases Adamts1, metalloproteinase-2 (Mmp2), and Hyaluronidase-2 (Hyal2). In contrast, Notch signaling from endocardial cells exerts the opposite function on these genes. Notch1 receptor binding by its ligand delta-like 4 (Dll4), induces cleavage of the Notch1-intracellular domain (N1ICD), which then translocates to the nucleus and acts as a co-activator of the Recombination signal binding protein for immunoglobulin kappa J region transcription factor (Rbpj) (Fig. 2) (Del Monte-Nieto et al. 2018). Consequently, increased surface tension due to the blood flow thrusting onto the ventricular apex leads to increased Nrg1/ErbB2 signaling in laminar trabecular CMs closer to the apex, as well as Dll4/Delta signaling in the overlaying endocardial cells. In turn, laminar trabeculae upregulate the synthesis of cardiac jelly genes, whereas endocardial cells respond by secreting metalloproteases (Fig. 2). As a result, endocardial cells begin excavating the cardiac jelly until they touch-down on the myocardial layer, whereas the laminar trabeculae strive to maintain the volume of cardiac jelly surrounding them (Fig. 2). These two processes will synergistically promote the formation of multiple endocardium-cardiac jelly cysts along the apicobasal axis of the ventricular lumen. Each cyst encloses a single laminar trabecular unit and nurtures its growth, either by promoting its clonal expansion or by incorporating additional CMs extruding from the compact layer (Fig. 2) (Del Monte-Nieto et al. 2018).
Compared to zebrafish, trabeculation in mice is mediated by changes in CM metabolism. However, metabolic compartmentalization in mouse embryos is reversed compared to the developing zebrafish heart, with the compact myocardial layer exhibiting enhanced glycolysis relative to trabeculae (Guimaraes-Camboa et al. 2015). This effect is thought to be partly regulated by Hif-1α, the activity of which becomes compartmentalized in the compact zone and the developing interventricular septum, while it is inhibited in the trabeculae (Fig. 2) (Guimaraes-Camboa et al. 2015; Menendez-Montes et al. 2016). Stabilization of Hif-1α leads to its nuclear translocation, where it directly binds on hypoxia response elements present within regulatory sequences of glycolytic genes, such as hexokinase-1 (Hk1) and pyruvate kinase-M2 (Pkm2) and activates transcription (Fig. 2). For example, inactivation of Hif-1α in Nkx2.5-expressing cardiac progenitors and their descendants, by introducing an Nkx2.5-Cre driver in compound heterozygous mice carrying one floxed and one nullified Hif-1α allele, resulted in impaired induction of glycolysis and, consequently, diminished proliferative activity in compact CMs. As a result of the perturbed metabolic compartmentalization and proliferative defects, mice exhibited reduced compact myocardial wall thickness and interventricular septum defects and died in utero by E17.5 (Guimaraes-Camboa et al. 2015). Notably, Hif-1α has been suggested to enhance cell-cycle activity of compact CMs by directly repressing the expression of the tumor suppressor genes Cdkn1a, Cdkn1c, and Tob2, as well as by repressing the activities of p53 and the ATF4 cellular stress response pathway (Guimaraes-Camboa et al. 2015). Likewise, early, ectopic activation of Hif-1α through the introduction of a von hippel lindau (VHL)-floxed allele in Nkx2.5-Cre expressing cardiac progenitors resulted in ectopic activation of glycolysis in the trabecular layer and, consequently, impaired trabecular morphogenesis and conduction system development, myocardial wall thinning, IVS defects, ventricular dilation, and embryonic death by E17.5 (Menendez-Montes et al. 2016).
Interestingly, mice with a ventricular CM-specific knockout of Nkx2.5 also exhibit lethal trabeculation and conduction system defects, and this is likely attributed to a negative feedback loop between Nkx2.5 and Bmp10/Smad signals (Fig. 2) (Pashmforoush et al. 2004; Prall et al. 2007). Specifically, loss of Nkx2.5 leads to ectopic expression of Bmp10 throughout the developing ventricle, and, consequently, these mice exhibit hypertrabeculated ventricles due to aberrant trabecular CM proliferation, reminiscent of left ventricular non-compaction cardiomyopathies (Pashmforoush et al. 2004). Hence, Nkx2.5 balances the rate of proliferation, differentiation, and relative size of the trabecular and compact myocardial compartments by directly controlling Bmp10 expression (Fig. 2). Intriguingly, as Hif-1α has been found to directly bind and activate the expression of Nkx2.5, it is reasonable to speculate that Hif-1α acts as the direct upstream controller for the spatiotemporal compartmentalization of the Nkx2.5 and Bmp10 activities during trabeculation and compaction (Fig. 2) (Guimaraes-Camboa et al. 2015; Menendez-Montes et al. 2016; Yuan et al. 2017).
The compartmentalized activation of glycolysis by Hif-1α that drives the growth of new CMs in the compact myocardial layer is thought to be the result of hypoxic signaling (Guimaraes-Camboa et al. 2015; Menendez-Montes et al. 2016). However, thus far, direct evidence supporting the presence of hypoxia in these Hif-1α-active ventricular chamber domains has not been clearly established. For example, studies with the hypoxic probe pimonidazole have attempted to identify hypoxic regions (<2% oxygen) in the heart during development. Intriguingly, although the immunohistochemical co-localization of pimonidazole with nuclear Hif-1α signals has been clearly demonstrated in early CMs up to E9.5, most studies indicate separation of the two markers from ~E12.5 onwards. Specifically, at E12.5, pimonidazole is primarily localized in the outflow tract region, whereas Hif-1α nuclear immunoreactivity is exclusively detected in compact zone and interventricular septum CMs (Lee et al. 2001; Ream et al. 2008; Guimaraes-Camboa et al. 2015; Menendez-Montes et al. 2016). Thus, further studies will determine whether Hif-1α-mediated induction of glycolysis and proliferative expansion of compact CMs in the developing mouse heart is the result of hypoxia or perhaps aerobic stabilization of Hif-1α, as shown during cancer development (Luo et al. 2011).
Animal models of cardiomyocyte regeneration
Cardiomyocyte regeneration in fish and amphibians
A teleost fish, zebrafish (Danio rerio), is the best-studied lower vertebrate that can regenerate myocardium as an adult. Poss et al. (2002) demonstrated that up to 20% of the zebrafish cardiac ventricle can be surgically amputated and perfectly regenerate its original size, anatomy and function within 2 months post injury (Poss et al. 2002). A similar cardiac regenerative capacity has since been observed in adult newt and axolotl urodele, but not anurans, as well as in response to different types of damage such as cryoinjury (Nguyen et al. 2021). In contrast to surgical amputation, cryoinjury is closer to myocardial injury in mammals, as the initial stages of the injury response include thrombosis, accumulation of fibroblasts and collagen deposition (Bise et al. 2020). Remarkably, zebrafish can regenerate their hearts continuously. Bise et al. (2020) demonstrated that a previously cryoinjured, regenerated myocardium can be cryoinjured and regenerated again, and this may be repeated every month up to six consecutive times without exhausting its CM regenerative capacity, albeit the efficiency of scar resorption decreases with each cycle (Bise et al. 2020). The spectacular regenerative capabilities of the adult zebrafish heart are further demonstrated through a genetic experimental system of extreme myocardial injury, where >60% of the adult ventricular CMs are destroyed through Cre/loxP-mediated conditional expression of the diphtheria toxin gene (DTA). The massive loss of CMs disrupted electric conduction and elicited signs of severe cardiac failure, yet animals were able to fully regenerate and recover within several days (Wang et al. 2011).
A major reason to investigate these model organisms is to gain understanding of the origin and developmental potential of the cells that give rise to the regenerated CMs. To this end, zebrafish offer a robust experimental system due to their widespread and relatively straightforward application in transgenic, genome engineering and advanced imaging approaches. Most studies indicate that CM regeneration in these species occurs via epimorphosis: new myocardium expands proliferatively from pre-existing CMs under an apical epicardial cap that grows over the wound surface (Sallin et al. 2015). However, the type of epimorphosis seems to vary depending on the animal and injury model. For example, Cre/LoxP-based genetic fate-mapping and clonal analyses in zebrafish expressing transgenic constructs such as the Brainbow multicolor lineage tracer, indicating that the amputated ventricle regenerates via an organ-wide, stem cell-independent compensatory mode of epimorphosis. Specifically, a layer of subepicardial CMs known as the cortical myocardium, which normally envelope the whole adult ventricle, is activated within a week after the amputation and upregulates regulatory sequences for the Gata4 cardiac transcription factor (Fig. 3) (Gupta and Poss, 2012). Upon activation, cortical CMs de-differentiate, re-enter the cell cycle and migrate under the apical epicardial cap, into the trauma, before acquiring new positional values in a distal-to-proximal direction relative to the cut site (Gupta and Poss, 2012). Notably, the direction of regeneration in the amputated heart is the opposite of that seen in salamanders or in other regenerating zebrafish tissues like the fin, which are predominantly regenerated in a proximo-distal direction relative to the cut site (Gupta and Poss, 2012). More importantly, because the cortical layer of CMs is produced in zebrafish after birth from a small number of trabecular CMs that penetrate the compact layer and expand clonally under the ventricular surface (Gupta and Poss, 2012), these findings suggest that the source of CMs in the zebrafish heart changes upon transition from embryonic to the adult life (Fig. 3).
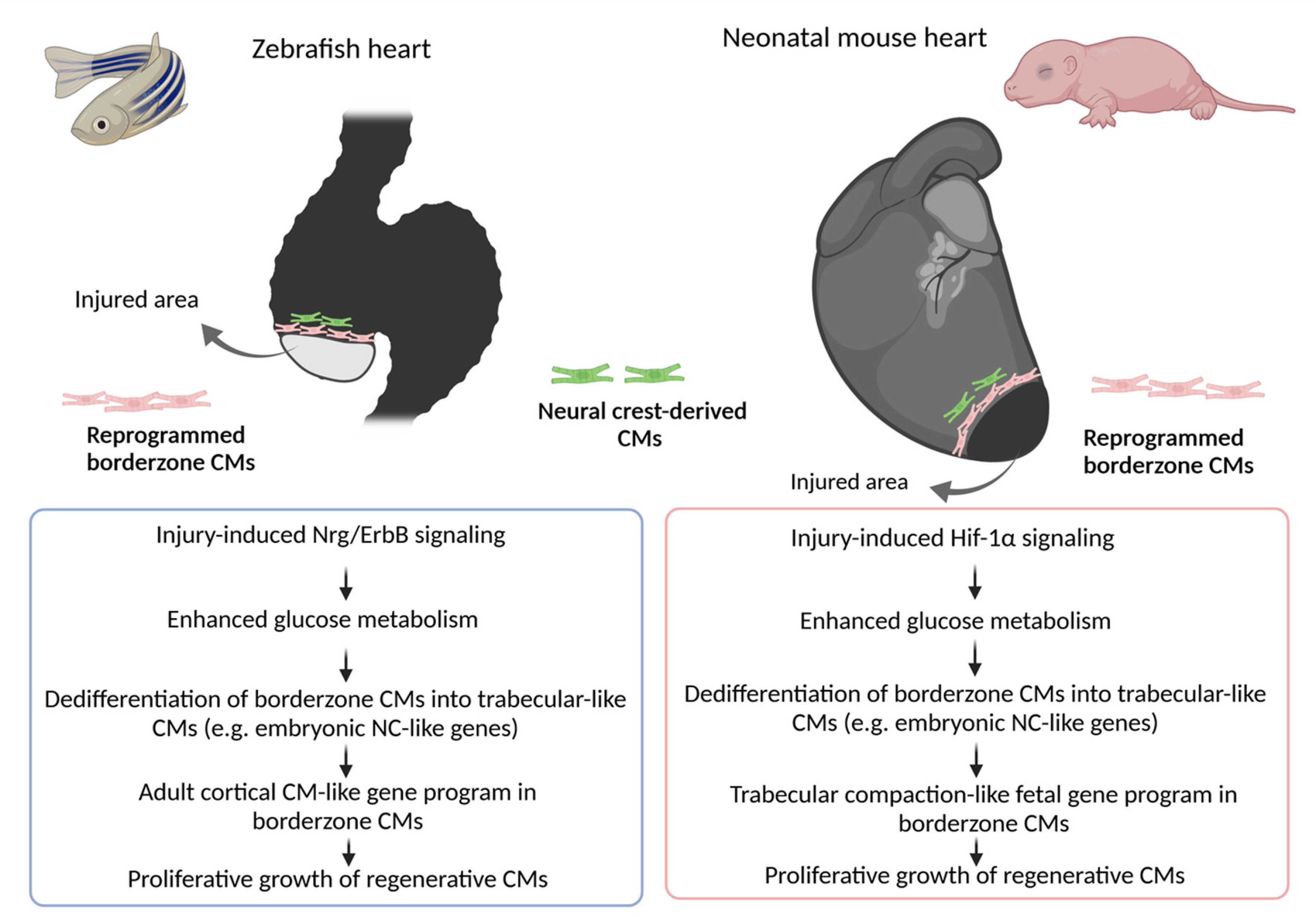
Fig. 3. Current models of cardiomyocyte regeneration in zebrafish and neonatal mice after heart injury.
CMs; cardiomyocytes, NC; neural crest.
On the other hand, regeneration of cryoinjured zebrafish myocardium is reported to follow a different pattern of epimorphosis that recapitulates aspects of embryonic development. Specifically, cryoinjury stimulates the transient formation of a blastema-like mass of undifferentiated, lineage-restricted cardiomyoblasts. These cardiomyoblasts are reprogrammed from spared CMs at the wound border by reactivating developmental genes such as Nppa and Tbx5a, which normally regulate the cardiac wall morphogenesis GRN that produces and patterns the trabecular and compact layers of the zebrafish myocardium during embryogenesis (Fig. 3) (Sallin et al. 2015). Subsequently, the blastema-like cells start to divide, and over the course of ~2 months, as the wounded heart regenerates, will re-differentiate into fresh and perfectly patterned myocardium by acquiring positional values in a proximo-distal direction relative to the wound border (Sallin et al. 2015; Sanchez-Iranzo et al. 2018; Sande-Melon et al. 2019). Notably, this type of blastema-dependent epimorphic regeneration is also seen in the amputated zebrafish fin, as well as during limb and heart regeneration in salamanders (Sallin et al. 2015).
Importantly, in both the amputated and cryoinjured zebrafish hearts, the cells that give rise to the regenerated CMs seem to deploy a developmental cardiac NC-like GRN (Fig. 3) (Sande-Melon et al. 2019; Tang et al. 2019). Specifically, genetic fate-mapping experiments indicate that the adult zebrafish heart is invested with a small pool of NC-CMs in the ventricle. These NC-CMs express regulatory sequences of Sox10 gene, which encode a NC-specific transcription factor. Upon injury, Sox10+ cells expand to a higher degree than other cardiomyoblasts and become an indispensable source of regenerated CMs; consequently, transgenic fish, in which adult Sox10+ ventricular cardiomyoblasts are destroyed through Cre/loxP-mediated conditional expression of DTA, exhibit incomplete heart regeneration (Sande-Melon et al. 2019). Notably, the NC comprises an important developmental source of trabecular CMs during embryonic cardiac wall morphogenesis in zebrafish (Fig. 2); and NC-derived trabecular CMs have been shown to contribute to the postnatal development of the cortical myocardium (Sande-Melon et al. 2019, Tang et al. 2019). Thus, regeneration in the zebrafish heart involves the recapitulation of developmental programs that produce the trabecular and cortical myocardial layers (Fig. 3).
Another important characteristic of CM regeneration in zebrafish is the dependence on nerve supply (Mahmoud et al. 2015). Nerve-dependence was described in salamander limb regeneration almost two centuries ago, and is thought to be imposed on the limb only after nerves grow into it during embryonic development (Farkas and Monaghan, 2017). Strikingly, early limb development does not depend on nerves, and if innervation of the developing limb is prevented from happening at an early stage, then adult salamanders grow with so-called “aneurogenic” limbs, which can regenerate perfectly well in nerve-independent fashion (Farkas and Monaghan, 2017). This seemingly paradoxical phenomenon has been attributed to the concentrations of mitogens such as the newt anterior gradient protein (nAG), neuregulin-1 (Nrg1) and nerve growth factor (Ngf), which are produced in the developing limb before nerves grow in and assume control of their expression (Farkas and Monaghan, 2017). In a landmark study, Kumar et al. found that newt limb regeneration does not actually depend on the nerve axons themselves, but on the growth factor nAG, which is secreted by the Schwann cells wrapped around them. Secreted nAG binds to its receptor Prod1 on the surface of the blastema cells and induces proliferation. Prod1 is produced in a graded fashion along the proximo-distal axis of the wound, in response to RA signaling. Consequently, regeneration in denervated limbs could be fully rescued by exogenous nAG expression (Kumar et al. 2007). In the zebrafish heart, Mahmoud and colleagues recently found that disruption of cholinergic, but not adrenergic, nerve supply inhibits proliferation and, thereby, curtails regeneration (Mahmoud et al. 2015). Notably, nerves are not required during embryonic cardiomyogenesis, suggesting that, as in the case of salamanders, nerve-dependence is likely imposed after the nerves grow into the zebrafish heart muscle (Mahmoud et al. 2015). Importantly, however, transgenic fish with a 90% reduction in cardiac innervation due to transgenic myocardial overexpression of semaphorin3aa are grossly normal but exhibit impaired cardiac regenerative capacity (Mahmoud et al. 2015). Thus, it remains unclear whether nerve-dependence can be bypassed in the adult regenerating zebrafish heart.
Cardiomyocyte Regeneration in mammals
Although much of our current understanding of cardiac regenerative biology is the result of experimenting with fish and amphibians, both animal models have important evolutionary differences compared to mammals. For example, the heart and circulatory systems in these species are fundamentally different from the latter. The cold-blooded zebrafish are equipped with a two-chambered heart -a spongy, highly trabeculated ventricle and an atrium- that mainly pumps deoxygenated blood from the sinus venosus (inflow tract) into the bulbus arteriosus (outflow tract) before it becomes oxygenated in the gills and return to the body (Poss et al. 2002). In contrast, in placental mammals, including mice and humans, the heart becomes developmentally patterned into four chambers, so that oxygenated and deoxygenated blood flows are completely separated after birth to the left and right sides, respectively. Meanwhile, the trabecular myocardium becomes compacted into the ventricles and the interventricular septum that will divide them (Del Monte-Nieto et al. 2018; Hatzistergos et al. 2020). Thus, although many of the molecular and cellular mechanisms underlying cardiac regenerative biology may be conserved between different classes of animals, there are several mammalia-specific developmental and regenerative processes that cannot be fully investigated in fish and amphibians.
A groundbreaking study by Porrello et al. (2011) provided an unprecedented opportunity to understand heart regeneration in mammals, by showing that, like zebrafish, 1-day-old (PN1) neonatal mice can also regenerate their heart after surgical resection, cryoinjury, or experimental MI (Porrello et al. 2011; Sadek and Porrello, 2020). The exact time when mice lose their cardiac regeneration capability, referred to as regenerative window, is not clear yet. Some studies indicate that the regenerative window closes before the end of the first week post-birth, as PN7 mice are not able to regenerate their heart and respond with cardiac fibrosis; whereas others suggest that it is abolished by ~48 hours (Sadek and Porrello, 2020). Of note, there are also few reports refuting the capacity of the neonatal mouse heart to regenerate (Sadek and Porrello, 2020); or arguing that the efficiency of scar resorption may vary depending on the size and type of injury, as well as on confounding factors such as differences between mouse strains, housing conditions, methodological and operator variability (Sadek and Porrello, 2020).
On the other hand, emerging evidence suggests that transient cardiac regenerative potential might also be present in large mammals, such as newborn pigs. The pig heart is anatomically and physiologically closer to humans, and therefore pigs are an outstanding translational animal model for the study of cardiac regenerative medicine. Recent work has demonstrated that within the first 48 hours of birth, newborn pigs can regenerate their myocardium after an experimentally induced heart attack, but this capacity disappears in 3-day-old animals (Sadek and Porrello, 2020). Whether a similar cardiac regenerative window exists in newborn humans remains unclear. Support for this exciting hypothesis may be drawn from cases of complete functional myocardial infarction (MI) in newborn children; as well as from the observation of limited or no post-operative scarring for the rare congenital heart disease anomalous left coronary artery from the pulmonary artery (ALCAPA) in newborns (Sadek and Porrello, 2020).
Compared with fish and amphibians, a mammalian model organism can provide better understanding of the adult origin and developmental potential of the cells that give rise to the regenerated CMs in humans. Consequently, neonatal mice have become the primary animal model because, compared with large mammals, they are easy to house and breed; have a relatively brief life-cycle; have relatively large litters; and, most importantly, are widely popular in genetic experiments .
Accordingly, fate-mapping experiments suggest that, as in the case of zebrafish and urodeles, CM regeneration in newborn mice is stem cell-independent and occurs via an organ-wide compensatory mode of CM proliferation (Sadek and Porrello, 2020). For example, in their original study, Porrello et al. (2011) employed a genetic fate-mapping approach whereby a Rosa26-stop-lacZ reporter was combined with a tamoxifen-inducible αMHC-MerCreMer construct to irreversibly tag neonatal CMs with β-galactosidase before surgical amputation. Although this initial approach to lineage-trace the regenerative cells exhibited limited sensitivity, since ~30-40% of newborn CMs eluded Cre-mediated β-galactosidase tagging, the researchers made several important observations that helped them conclude that regenerated CMs are more likely to be amplified from pre-existing ones. First, they recorded an organ-wide increase in CM cell-cycle activity, which peaked at 7 days post-injury. Second, at 21 days post amputation, most of the regenerated CMs carried the β-galactosidase genetic tag, thereby arguing against a stem cell-dependent mechanism that theoretically would be expected to yield mostly β-galactosidase negative CMs (Porrello et al. 2011). In another study, Li et al. (2019) attempted to overcome the lineage-tracing limitations by developing a more sensitive, dual-recombinase, intersectional genetic fate-mapping system involving a Tnnt2-Cre; a tamoxifen-inducible Rosa26-DreER; and a double-fluorescence reporter which expresses green or red fluorescent lineage-tracers in response to Cre or Dre recombination, respectively (Li et al. 2019). Compared with Porrello et al. (2011) this approach enabled the researchers to permanently tag most, if not all, Tnnt2-Cre-expressing newborn CMs with green fluorescence; while non-myocytes, including presumptive stem cells, could be marked with red fluorescence in response to tamoxifen-induced embryonic activation of the Rosa26-DreER construct. The study reported that apical resection in neonatal pups carrying the dual recombinase system was accompanied by expression of green but not red fluorescence lineage tracers in regenerating CMs. Therefore, consistent with Porello et al. (2011), it was concluded that regenerated CMs are progeny of the spared CMs, rather than being de novo produced from stem cells (Li et al. 2019). However, despite the use of highly sophisticated genetic tools, a few potentially important limitations of the experimental design should be noted. For example, although CM regeneration in neonatal mice takes at least 21 days to complete, the study was terminated at 14 days after amputation, and therefore a potential contribution from stem cell-dependent mechanisms at later stages of regeneration was not addressed. Moreover, whether the regenerated CMs were produced through a blastema-like or compensatory epimorphic regeneration could not be addressed, since Tnnt2-Cre expression irreversibly activates the green while neutralizing the red fluorescence lineage tracer in recombined cells. Finally, tamoxifen-induced recombination of the Rosa26-DreER construct was performed during embryonic development, at either embryonic day (E12.5) or E16.5, but whether this was sufficient to label most non-myocytes and putative stem cells in the neonatal heart, was not clarified.
In fact, other studies suggest blastema-like epimorphic regeneration mechanisms may be in effect in the regenerating newborn mouse heart, like in zebrafish. For example, Jesty et al. (Jesty et al. 2012) and Konfino et al. (Konfino et al. 2015) reported that CM regeneration of cryoinjured and amputated neonatal murine myocardium, respectively, involves the upregulation of the proto-oncogene c-Kit in proliferative cardioblasts at the border of the wounded myocardium. An intersectional genetic-fate mapping study utilized the Cre/LoxP and FlpE/Frt recombinase systems and found that c-Kit is also upregulated in a subset of NC cells that contribute trabecular CMs during mouse embryonic development (Hatzistergos et al. 2015). As with zebrafish, a small number of NC-derived cardioblasts persist in the postnatal murine myocardium and may be guided to migrate and differentiate in vitro by SDF1/CXCR4 signaling (Hatzistergos et al. 2015; Hatzistergos et al. 2016; Hatzistergos et al. 2020). Intriguingly, Sdf1/Cxcr4 signaling is also involved in blastema formation during mammalian digit tip regeneration, and its disruption curtails CM proliferation and regeneration in both the adult zebrafish and neonatal mouse heart, although whether this defect relates to the NC-derived CMs is not clear (Das et al. 2019).
Notably, the role of c-Kit as a marker of regenerating cells in the neonatal mouse heart remains debated, as a recent genetic fate-mapping study failed to detect significant upregulation of c-Kit regulatory sequences in regenerating CMs (Elhelaly et al. 2019). However, a possible explanation of these seemingly discrepant results may be found to the modularity of the c-Kit gene regulatory elements: in the latter animal model, c-Kit sequences that drive reporter gene expression seem to be active mostly in endothelial rather than neuronal/NC cell types (Elhelaly et al. 2019), whereas the opposite expression pattern is observed with different c-Kit DNA sequences (Hatzistergos et al. 2015). Similarly, the neonatal mammalian heart has been shown to retain a small pool of cardiomyoblasts expressing the transcription factor Isl1, and earlier studies suggested that these postnatal Isl1+ cardiomyoblasts are likely aSHF-derivatives. However, recent work employing a combination of single-cell transcriptomics and conventional lineage tracing experiments in mice, clarified that these cells are NC derivatives that express Isl1 through neural rather than aSHF-specific regulatory sequences, and contribute to the cardiac wall morphogenesis program during embryonic development (Hatzistergos et al. 2020).
As in the case of salamanders and fish, regeneration of neonatal mouse CMs depends on nerve supply, while embryonic development does not. White et al. (2015) utilized a Wnt1-Cre fluorescent reporter mouse line to image the growth of the cardiac sympathetic nervous system from NC cells into the neonatal mouse heart (White et al. 2015). They observed that surgically amputated 2-day old hearts responded with heavy dendrite hyper-innervation at the wound border, which peaked at 2 weeks post-injury. By 21 days, the amputated ventricle was fully regenerated and innervated by sympathetic axons. Attenuating innervation of the wounded ventricle through chemical sympathectomy of adrenergic nerves with 6-Hydroxydopamine hydrobromide curtailed the regenerative process (White et al. 2015). Similarly, Mahmoud et al. (2015) subjected 1-day old neonatal mice to surgical amputation or experimental MI and found that chemical or mechanical denervation of parasympathetic cholinergic nerves resulted in reduced CM proliferation and impaired regeneration (Mahmoud et al. 2015). Collectively, the two studies suggest that, as with salamander limbs (Farkas and Monaghan, 2017), heart regeneration in mammals is nerve-dependent, and that it is probably the amount rather than the type of innervation that regulates the regenerative process.
Notably, Mahmoud et al. (2015) reported that nerve dependence in the regenerating neonatal mouse heart could be partly bypassed by exogenous Nrg-1 expression (Mahmoud et al. 2015), a finding reminiscent of denervated axolotl and newt limb regeneration rescue experiments (Farkas and Monaghan, 2017). However, whether Nrg-1 is directly secreted by intracardiac nerves or other nerve-dependent regenerative cells, remains unclear. In fact, previous work in zebrafish identified a pool of injury-activated, trabecular CMs and epicardium-derived perivascular cells as the main sources of Nrg-1; secreted Nrg-1 then binds to ErbB2/4 receptors on the surface of cortical CMs to activate intracellular signals that enhance cell-cycle re-entry (Harvey et al. 2016). As previously discussed, both mice and zebrafish activate cardiac Nrg-1/ErbB signaling to control the development of trabecular CMs during the embryonic cardiac chamber morphogenesis program (Harvey et al. 2016; Fukuda et al. 2019; Honkoop et al. 2019). Therefore, these findings are consistent with previous observations that zebrafish evoke developmental programs related to trabecular CM growth and differentiation, for cardiac regenerative purposes (Fig. 3) (Sanchez-Iranzo et al. 2018; Fukuda et al. 2019; Honkoop et al. 2019; Sande-Melon et al. 2019; Tang et al. 2019). In mice however, Erbb2 and Erbb4 CM receptors undergo rapid and dramatic downregulation upon closure of the postnatal cardiac regenerative window (Harvey et al. 2016). Consequently, mice carrying a constitutively active CM-specific ErbB2 transgene exhibit a prolonged cardiac regenerative window, albeit they subsequently develop cardiomegaly and lethal hypertrophic cardiomyopathy, presumably due to uncontrolled neonatal CM proliferation (Harvey et al. 2016). Notably, whether loss of early neonatal Nrg-1/ErbB signaling from CMs and/or intracardiac nerves is sufficient to inhibit regeneration has not been experimentally tested.
In zebrafish embryos, Nrg-1/ErbB signaling controls the development of trabecular CMs by inducing a “glycolytic switch”. Nrg-1/ErbB signals the prospective trabecular CMs to increase glucose utilization as their metabolic substrate while upregulating the expression of glycolytic genes such as Hk1 and Pkm2, at the expense of mitochondrial respiration. Surprisingly, the switch to glycolysis does not induce proliferation but activates an EMT-like process, causing the CMs to undergo apical constriction of their cytoskeleton, delaminate and seed the trabecular layer (Fukuda et al. 2019). Through a combination of conventional and single-cell transcriptomic lineage-tracing experiments, Honkoop et al. (2019) found that a similar trabeculation-like glycolytic switch triggered by Nrg-1/ErbB2 is required for regeneration of the cryoinjured zebrafish myocardium. However, compared with trabecular CM development, injury-induced Nrg-1/ErbB2 acts specifically in regenerative CMs proximal to the wound border, triggering their dedifferentiation, migration, as well as cell cycle re-entry (Fig. 3) (Honkoop et al. 2019). Notably, Hif-1α is a master glycolytic transcription factor that directly binds and activates pro-glycolytic genes, including Hk1 and Pkm2 (Luo et al. 2011). Although previous studies suggest that transient, hypoxia-induced Hif-1α signaling is required for zebrafish CM dedifferentiation, proliferation, and regeneration (Sadek and Porrello, 2020); Honkoop et al. (2019) argue that the Nrg-1/ErbB signaling-mediated cardiac glycolytic switch occurs independently from Hif-1α (Honkoop et al. 2019).
The murine embryonic cardiac chamber morphogenesis program is also dependent on compartmentalization of the myocardial metabolism. Interestingly, the metabolic program of the developing mouse heart more closely matches the regenerating than the developing fish heart. During ventricular trabeculation and compaction, the glycolytic switch becomes activated in the proliferating compact myocardial layer, while development of the non-proliferating trabeculae relies on mitochondrial oxidative metabolism (Guimaraes-Camboa et al. 2015; Menendez-Montes et al. 2016). These regional differences in mouse CM metabolism are thought to be regulated by oxygen tension, which leads to compartmentalized stabilization or inactivation of Hif-1α and its downstream glycolytic GRN in the compact and trabecular myocardial layers, respectively (Guimaraes-Camboa et al. 2015; Menendez-Montes et al. 2016). Remarkably, recent work by Sadek and colleagues suggests that the progressive loss of the cardiac regenerative window in mice may be explained by the transition of mouse embryos to the oxygen-rich postnatal environment (Sadek and Porrello, 2020). This leads to a permanent metabolic remodeling in CMs, whereby fatty-acid β-oxidation in the mitochondria dominates over Hif1α-mediated utilization of glucose as the metabolic substrate. Consequently, during the cardiac regenerative window, neonatal mice evoke this Hif-1α mediated glycolytic developmental program to induce de-differentiation, migration and proliferative growth of border zone CMs (Fig. 3) (Sadek and Porrello, 2020). In contrast, closure of the cardiac regenerative window coincides with oxidative stress and subsequent accumulation of reactive oxygen species (ROS), oxidative DNA damage, DNA damage response (DDR) markers and polyploidization of neonatal CMs (Sadek and Porrello, 2020). Accordingly, exposure of neonatal mice to hyperoxemia and ROS generators shortens the regenerative window; whereas several experimental interventions such as exposure to gradual systemic hypoxemia, DDR inhibition, ROS scavenging or inhibition of fatty-acid utilization may prolong it, by delaying the cell-cycle arrest and polyploidization of postnatal CMs (Sadek and Porrello, 2020).
The crosstalk between cardiac metabolism and CM proliferation in the regenerating heart seems to converge on Hippo signaling, an evolutionarily conserved pathway that, during development, acts as a mechanosensing mechanism to control organ-size, including the mouse heart (Wang et al. 2018). Increased surface tension -for example due to increased blood flow, cell-matrix stiffness, CM contractility or overcrowding of proliferating cells- activates Hippo signaling, which in turn marks its two nuclear effectors Yap and Taz for proteasomal degradation through the Lats1/2 kinases (Wang et al. 2018). In the absence of Hippo signaling, Yap and Taz translocate to the nucleus and interact with the Smad family members TEADs (Transcriptional Enhanced Associate Domain), as well as with Hif-1α, to induce transcription of cell-cycle and glycolytic genes, including Pkm2 (Wang et al. 2018). In the mouse heart, Nrg1/Erbb2 signaling activates Yap through an Erk-dependent mechanism that leads to non-canonical Yap phosphorylation on S274 and S352 (Aharonov et al. 2020). Moreover, Nrg1/Erbb2 signaling induces cytoskeletal changes in CMs that, for example, lead to the release of the scaffold protein Angiomotin-like 1 (Amotl1) from adherent junctions. Upon release, Amotl1 interacts with and activates Yap and, together, they bypass the Lats1/2 kinases and translocate to the nucleus (Wang et al. 2018). In breast cancer cells, Nrg1 binding to ErbB4 leads to cleavage of the ErbB4 intracellular domain, which in turn is released into the cytoplasm and interacts with Yap to bypass its Lats1/2-mediated inactivation and promoting its nuclear translocation (Wang et al. 2018). However, whether Nrg1-ErbB4-Yap signaling is also involved in CM proliferation and regeneration is currently not known.
Yap-induced proliferation and metabolic remodeling of regenerating neonatal mouse CMs has also been shown to require activation of Toll-like receptor signaling by the immune cells that infiltrate the wound (Wang et al. 2018). Similarly, cell-matrix adhesions can also regulate CM metabolism and proliferation in the regenerating heart. Bassat et al. identified Agrin, a proteoglycan involved in neuromuscular junction development, as a critical cardiac ECM component of the regenerating neonatal mouse heart (Wang et al. 2018). Agrin binds to the dystrophin–glycoprotein complex on the surface of CMs, leading to inhibition of Hippo pathway and Yap translocation to the nucleus, thereby inducing CM proliferation. Interestingly, MesP1cre+/−;Agrnfl/fl mice, in which Agrin has been conditionally deleted in Mesp1 cardiac progenitors and their derivatives, develop normally, but regeneration following apical amputation is less robust compared with controls (Wang et al. 2018). Therefore, further investigation into the role of Agrin in heart development and regeneration is warranted. Recently, Liu et al. (2020) demonstrated that cardiac lymphatic endothelial cells (LECs) produce Reelin (Reln), another ECM glycoprotein known for its role in neural cell migration and differentiation. Global or LEC-specific Reln mutant neonatal mice develop smaller hearts and exhibit impaired regenerative capacity, likely due to increased CM apoptosis and decreased proliferation (Liu et al. 2020). Previous studies on neural and cancer cell development and migration have identified that Reln-mediated cell-matrix adhesions are mainly established through the two members of low-density lipoprotein receptor gene family: the very-low density lipoprotein receptor (VLDLR) and the apolipoprotein E receptor 2 (ApoER2); but also through integrin-β1 (Liu et al. 2020). Intriguingly, Liu et al. (2020) provided evidence that β1 integrins in CMs are required for Reln to exert its effects on CM proliferation and survival signaling; although molecules such as disabled 1 (Dab1), Akt and Erk downstream of VLDLR and ApoER2 signaling, were also found to be upregulated. A similar Reln/Integrin-β1 mechanism has been shown to induce glycolysis and the Warburg effect in cancer cells, but whether the Reln effects on CM proliferation and survival are linked to metabolic remodeling is not known (Qin et al. 2017).
Development-guided therapeutic approaches for heart regeneration
Upon closure of the cardiac regenerative window, mammals are unable to replace damaged CMs with new, healthy ones, and consequently, any loss in myocardial tissue is replaced by fibrous scars, eventually leading to myocardial dysfunction, ventricular remodeling and HF. Currently, there is no therapy for heart diseases, and most treatments mainly aim at delaying cardiac remodeling. Several pharmacological approaches, including β-blockers (e.g., carvedilol), angiotensin-converting enzyme inhibitors (e.g., enalapril), angiotensin receptor-neprilysin inhibitors (e.g., LCZ696), as well as mineralocorticoid receptor antagonists (diuretics, e.g., spironolactone), have been successful in reducing mortality and morbidity. Many patients, however, eventually progress to end-stage HF and become dependent on mechanical support by left ventricular assistant devices and cardiac resynchronization therapy before requiring organ transplantation. Accordingly, there is an increasing need to develop novel approaches for cardiac regenerative medicine, with cell replacement therapies currently being touted as the most promising (Hashimoto et al. 2018; Menasche, 2018).
By studying cases of male patients who had received heart transplants from female donors, some researchers provided evidence of cardiac microchimerism, whereby rare CMs carrying the Y-chromosome could be detected by in situ hybridization within the female donor hearts (Eschenhagen et al. 2017). This simple yet paradigm-shifting observation opened the possibility that adult CM regeneration occurs in humans, albeit at low levels. The XY-chromosome CMs were interpreted as the result of circulating stem cells from the host that colonized the XX transplants and differentiated into new CMs, triggering frenzied research into identifying and therapeutically harnessing the most potent regenerative cardiac stem cell (Eschenhagen et al. 2017; Menasche, 2018). However, subsequent genetic studies in mice suggested that most of the chimeric CMs were likely due to cell-cell fusions between host and donor cells, rather than stem cell regeneration (Eschenhagen et al. 2017). Nonetheless, recent work utilizing highly sophisticated, carbon-dating approaches corroborates the plasticity and regenerative capacity of the adult human myocardium. Based on these studies, it is estimated that the annual rate of CM turnover ranges from 1% at the age of 20 to 0.3% at the age of 75. This is sufficient to regenerate <50% of CMs throughout a normal lifespan, but falls far from the CM production capacity required to fully regenerate a damaged heart before progressing into ventricular remodeling and HF (Eschenhagen et al. 2017). Accordingly, the goal of cardiac regenerative medicine is to develop novel pharmacologic, genetic, or cell-based therapies that could either stimulate endogenous cardiomyogenesis or supply the necessary amount of CM replacement cells exogenously (Fig. 4).
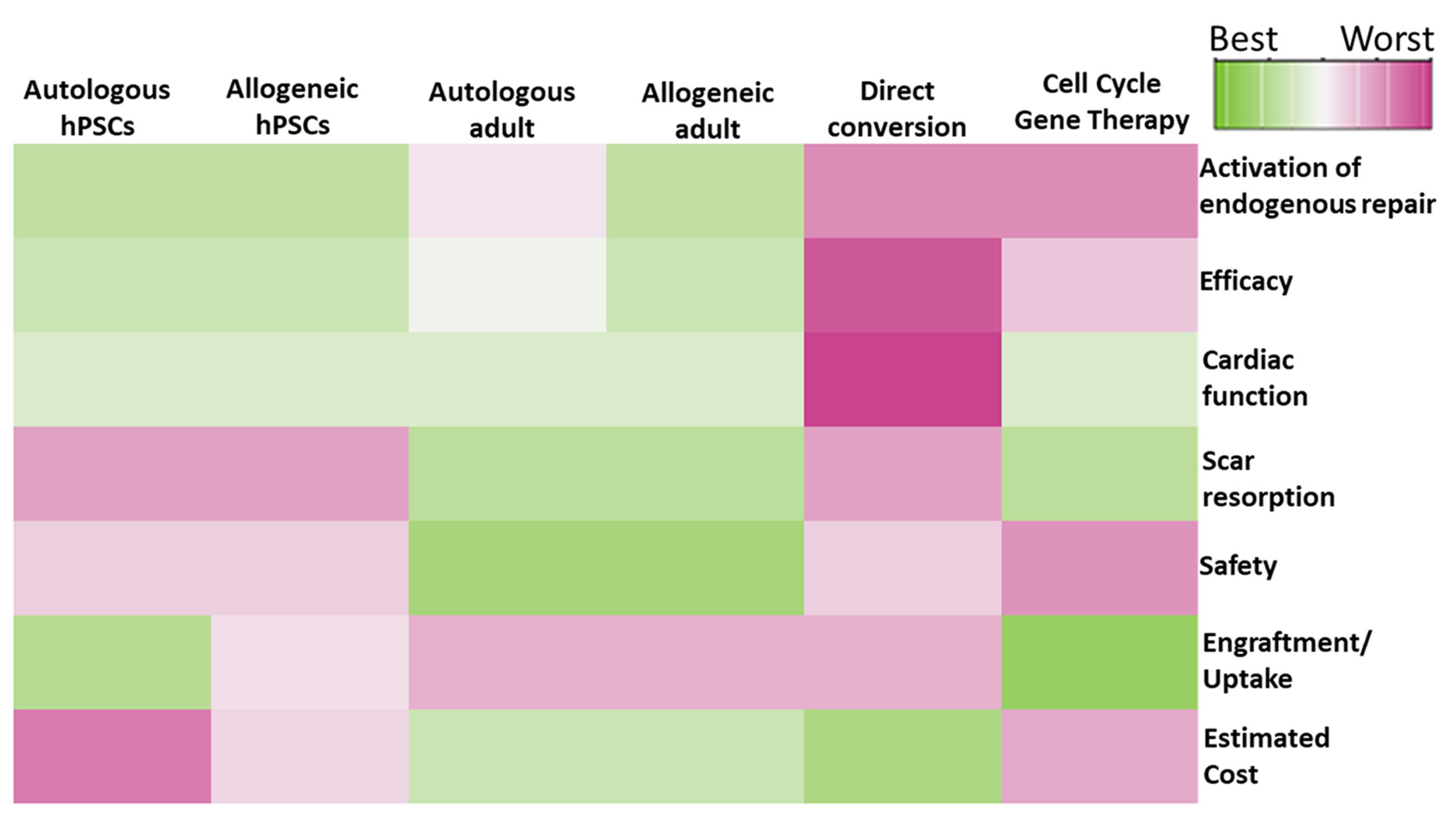
Fig. 4. Therapeutic score heatmap.
Columns indicate therapeutic modalities, rows indicate metrics. Green and pink colors code for advantages and disadvantages, respectively. hPSC; human pluripotent stem cells.
Adult cell therapies
Among the first cell types that entered the clinical trials are autologous skeletal myoblasts. These are a highly proliferative myogenic cell types that can be readily obtained from muscle biopsies and expanded in vitro into large quantities, and were originally considered capable of directly re-muscularizing the failing heart (Eschenhagen et al. 2017; Menasche, 2018). In patients and preclinical animal models of ischemic HF, autologous skeletal myoblast therapy led to modest improvements in heart function. However, further studies concluded that any beneficial effects are probably indirect; for example, due to paracrine effects that lead to improved vascularization and reduction of fibrosis, since evidence for long-term engraftment and direct remuscularization have been minimal, at best. In addition, skeletal myoblasts are determined by the skeletal myocyte lineage and do not transdifferentiate into cardiac myocytes. Consequently, they fail to couple electromechanically with the host myocardium due to downregulated N cadherin and connexin 43 expression. As a result, patients receiving skeletal myoblast therapy are at risk of developing unwanted arrhythmias (Menasche, 2018). Nonetheless, skeletal myoblast transplantation, in the form of fibrin glue-reinforced cell sheets, sutured over the epicardium during open heart surgery, became the first cell-based therapy that received conditional and time-limited regulatory approval in Japan (Konishi et al. 2016).
As expected, early reports on chimerism of the transplanted heart from circulating cells propelled the interest in cell therapy studies with bone marrow cells, mostly in the form of freshly isolated, unselected mononuclear fractions or culture-expanded mesenchymal stem cells (MSCs) (Menasche, 2018). Following almost two decades of animal and clinical studies, a consensus has been reached that, as with skeletal myoblasts, bone marrow cell transplants neither engraft long-term nor transdifferentiate or re-muscularize the heart after damage. Therefore, any potential therapeutic benefit is probably indirect, due to transient cell-cell interactions with the host and secretion of paracrine factors (Eschenhagen et al. 2017; Menasche, 2018). For example, using female pigs as a sex-mismatched experimental model for acute MI cell therapy, a minimally invasive catheter-based approach for intramyocardial injection of male MSCs within and around the injured area was developed, which produced significant structural and functional improvements in the infarcted hearts (Hatzistergos et al. 2010). However, further analysis indicated that any therapeutic benefit was unlikely to have been due to direct re-muscularization of the host myocardium by male MSCs, since the incidents of Y-chromosome detection by in situ hybridization in CMs were rare (Hatzistergos et al. 2010). Strikingly, 2 weeks after MSC transplantation, a significant increase in the number of host CMs undergoing mitosis within and around the infarct site was noted, and this effect was accompanied by a dramatic upregulation in c-Kit expression within and around the infarct site (Fig. 4). (Hatzistergos et al. 2010). These findings are reminiscent of the mechanisms activated during endogenous CM regeneration in the neonatal mouse heart, including the activation of a NC cell-like developmental GRN in border zone cardiomyoblasts (Jesty et al. 2012; Konfino et al. 2015; Sande-Melon et al. 2019; Tang et al. 2019). Moreover, consistent with the cardiac regenerative mechanisms described in neonatal mice and adult fish, MSCs may stimulate endogenous repair by modulating glucose metabolism (Cai et al. 2016) and the immune responses in the host myocardium (Aurora et al. 2014; Vagnozzi et al. 2020). Considering these findings, a multi-center, double-blind, placebo-controlled, Phase II clinical trial recently tested a novel approach, whereby patients with ischemic HF received a combination therapy with culture-expanded autologous MSCs and heart biopsy-derived cKit-expressing cells, on top of their maximal guideline-directed therapy (Bolli et al. 2021). Although the therapy was not sufficient to reverse the chronic structural and functional deteriorations in terms of scar size and ejection fraction changes, patients experienced significant improvements in clinical outcomes, and in particular a reduction in major adverse cardiac events and improved quality of life (Bolli et al. 2021).
Notably, the expression of c-Kit as well as several other markers including Sca-1 and the transcription factor Isl1 in the adult heart have been highly contested, since they were touted as markers of bona fide postnatal cardiovascular (CV) stem cells that could be isolated, expanded ex vivo and used as cell replacement therapy for HF (Eschenhagen et al. 2017). However, several genetic lineage-tracing studies in mice have recently challenged this possibility (Hatzistergos et al. 2015; Eschenhagen et al. 2017; Hatzistergos et al. 2020).
Cell therapy with pluripotent stem cell derivatives
Most of the interest in cardiac regenerative medicine is currently focused on human pluripotent stem cells (hPSCs), in either their embryonic (hESCs) or lab-induced (hiPSCs) form (Eschenhagen et al. 2017; Yamanaka, 2020). Compared with somatic cells, which have restricted developmental potential and limited ability to differentiate into healthy CMs, hPSCs are pluripotent, meaning they are equivalent to the cells which form the epiblast of the early human embryo and, consequently, differentiate in vitro into tissues from all three germ layers -ectoderm, mesoderm, endoderm- including CMs (Eschenhagen et al. 2017; Yamanaka, 2020). The discovery and development of the proper, chemically defined in vitro conditions that enable the culture and expansion of hPSCs and their subsequent directed differentiation into bona fide CM-forming cells, provided the scientific community with an unprecedented capacity to grow theoretically unlimited amounts of spontaneously beating, bona fide CMs in the lab. This has led to a renewed enthusiasm for realizing therapeutic heart regeneration in humans, as lab-grown hPSC-CMs are considered the “holy grail” of direct CM replacement strategies (Eschenhagen et al. 2017; Yamanaka, 2020).
The choice between hESC- or hiPSC-derived CMs for clinical use depends on several factors, most significantly ethics, cost-effectiveness and immunogenicity (Fig. 4). For example, the derivation of hESCs depends on the destruction of human blastocysts, and this has been the subject of considerable ethical debate and regulatory hurdles that hampered stem cell research and regenerative medicine for years (Yamanaka, 2020). In addition, the choice of a hESC-based approach precludes the development of autologous CMs, thereby raising important concerns regarding the immunocompatibility and, hence, safety and efficacy of heterologous grafts (Yamanaka, 2020). On the other hand, hiPSC-based approaches can solve these issues but create others. For example, manufacturing of autologous hiPSC-CM grafts may not always be clinically feasible for several reasons (Fig. 4). First, reprogramming and validation of clinical-grade autologous hiPSC lines and the subsequent scaling-up and validation of hiPSC-CM manufacturing processes and end-products for each patient are currently major hurdles, as they can drastically increase the regulatory and economic burden (Yamanaka, 2020). In addition, an autologous hiPSC-CM graft cannot be readily available unless it is developed prospectively and cryopreserved in tissue banks, which again introduces important economic, regulatory, and social ramifications, since not every patient may be able to afford this treatment. Finally, autologous hiPSC-CMs may not be an optimal therapy for patients with inherited or congenital cardiomyopathies, unless the disease-causing genetic or epigenetic defects are identified and corrected prior to transplantation (Fig. 4).
Potential solutions to the above problems include the establishment of large hiPSC banks for HLA-matched tissue transplantation, or the development of universally immunocompatible mutant hiPSC lines that could be used for manufacturing off-the-shelf allogeneic grafts (Yamanaka, 2020). For example, Xu and colleagues (2019) demonstrated a novel approach, whereby CRISPR-Cas9-mediated selective inactivation of human leukocyte antigen (HLA) class-I molecules HLA-A/B, but not HLA-C, could prevent attack by CD8+ T and NK cells respectively and, therefore, graft rejection. A small bank of ~12 genetically engineered HLA-C-retained hiPSC lines is estimated to theoretically provide immunocompatible grafts for >90% of the world’s population (Xu et al. 2019). Further work in human clinical trials is expected to verify these estimations, as well as the oncologic risk and true levels of immune tolerance of such cells.
An off-the-shelf allogeneic iPSC-CMs cell therapy model was recently put to preclinical testing in non-human primates (Shiba et al. 2016). A single Philippine cynomolgus monkey with MHC haplotype homozygosity was identified and used as a universal iPSC-CM donor in five MHC-heterozygous animals. The heterozygous monkeys were subjected to experimental MI, and 2 weeks later underwent sternotomy to receive 400 million iPSC-CMs or vehicle, delivered via 10 trans-epicardial injections into the left ventricular infarct and border zones. Remarkably, the iPSC-CMs, which were tagged with the fluorescent calcium indicator G-CaMP7.09-GFP, engrafted and survived for at least 12 weeks after transplantation and became electrically coupled with the host. Crucially, despite the MHC heterozygosity, animals also required mild immuno-suppression throughout the study period to escape immune rejection. Interestingly, cardiac function was significantly improved compared to vehicle, but scar tissue persisted to comparable levels between the two groups. Moreover, four out of five iPSC-CM treated animals experienced sustained ventricular tachycardias -likely due to the immature phenotype of the cells, as well as undesired specification of some iPSC-CMs to atrial, right ventricular, or conduction system lineages- raising important concerns about the safety and efficacy of the model in patients with HF. Thus, despite the encouraging findings on engraftment and improved cardiac function, these results challenge the original hypothesis of direct scar tissue replacement by exogenously delivered iPSC-CMs. In fact, further studies suggest that PSC-based therapies might exert their beneficial effects indirectly, through paracrine actions. For example, Zhao et al. (2021) demonstrated in a xenogeneic porcine model of cell therapy post-MI that transgenic overexpression of cyclin D2 under the control of αMHC promoter sequence could significantly boost the limited effects of hiPSC-CM therapy in scar size reduction. Strikingly, the enhanced efficacy of cyclin D2-over-expressing hiPSC-CMs was likely due to enhanced proliferation of the engrafted cells, but most importantly due to the paracrine effects which stimulated endogenous regenerative mechanisms, as indicated by the enhanced proliferation of recipient CMs and vascular cells (Zhao et al. 2021).
As with adult cell therapies (Konishi et al. 2016; Bolli et al. 2021), other clinically feasible approaches that are currently being tested for improving safety, survival, engraftment, maturity, and paracrine effects of allogeneic hiPSC-CM therapies include combination therapy with fibroblasts, vascular cells or MSCs (Park et al. 2019), as well as bioengineering into 3D-cell sheets (e.g., clinicaltrials.gov, NCT04696328) (Menasche, 2018). Moreover, developmental biology-guided strategies are being developed to guide hPSC-CM specification away from unwanted, and potentially arrhythmogenic, CM lineages such as atrial and pacemaker CMs. For example, blockage of RA signaling during the stage-specific differentiation of hPSC-CMs prevents posteriorization of cardiogenic mesodermal progenitors into venous pole-like lineages and, therefore, favors specification mainly toward FHF and aSHF ventricular CMs, at the expense of atrial and pacemaker CMs (Lee et al. 2017). Further studies will determine the proper conditions for chamber-specific specification into left- or right-ventricular hPSC-CM lineages and whether these approaches may bring hPSC-based cardiac cell therapy closer to realization.
Gene and drug therapies
Apart from the quest for cell-based, regenerative cardiomyoplasty strategies, efforts are being made toward pharmacologic or gene therapies that can evoke endogenous CM regeneration (Fig. 4). A much vaunted approach has been the direct reprogramming of non-CMs to CMs (Miyamoto et al. 2018). The idea emerged as a breakthrough innovation following the original discovery on the induction of pluripotency in somatic cells by Yamanaka et al. and is targeted toward switching the fate of pro-fibrotic cells, like fibroblasts, which are abundantly present in the damaged heart, into regenerative CMs through the forced expression of an appropriate “gene cocktail” (Miyamoto et al. 2018; Yamanaka, 2020). In mice, a cocktail of three CM-GRN core transcription factors -Gata4, Mef2c and Tbx5 (or GMT)- has been identified as necessary and sufficient to induce the chromatin changes required for activating the CM-GRN in fibroblasts. When mouse embryonic fibroblasts are virally transduced in vitro with GMT-overexpressing constructs, ~10% of them convert into spontaneously beating CM-like cells. However, in vivo GMT gene therapy in adult mice post-MI diluted the initial enthusiasm, as less than 0.5% of the infiltrating fibroblasts convert into CM-like regenerative cells (Miyamoto et al. 2018). Interestingly however, although the amount of reprogrammed CM-like cells is far too low to be functionally important, mice receiving GMT gene therapy exhibit significant improvements in heart function and scar size reduction, suggesting that GMT therapy might exert additional, indirect therapeutic effects (Miyamoto et al. 2018). Moreover, since cardiac fibroblasts are indispensable for heart regeneration in zebrafish and neonatal mice (Sadek and Porrello, 2020), it is unclear whether achieving greater levels of reprogramming, at the expense of potentially exhausting the cardiac fibroblast pool, may be beneficial or detrimental for adult heart regeneration. In terms of cardiac reprogramming in humans, additional levels of complexity underlying the human cardiac GRN have rendered the GMT cocktail ineffective for converting human fibroblasts into CM-like cells (Miyamoto et al. 2018). A recent study suggested that the addition of 3 extra factors to the GMT cocktail -Mesp1, Myocardin and Mir133- is necessary, and could induce in vitro reprogramming of ~13% of adult human fibroblasts into CM-like cells. Furthermore, although reprogrammed human fibroblasts upregulate multiple CM-GRN genes, they do not exhibit full differentiation into spontaneously beating CMs, unless they are co-cultured with neonatal rat CMs (Miyamoto et al. 2018). Therefore, direct cardiac reprogramming is an exciting strategy, but more work is still needed prior to the clinical testing stage.
Other promising approaches include therapies targeted toward genes and proteins that have been found to regulate CM metabolism and proliferation. For example, low-fat diet, gene-knockdown of pyruvate dehydrogenase kinase 4-mediated fatty acid oxidation, or exposure to intermittent hypoxia to transiently enhance Hif-1α stability, have all been shown to augment glucose metabolism in adult mice; and thereby, recapitulate aspects of the metabolic switch that drives heart regeneration in fish and neonatal mice. Consequently, they may be potential stimulants of endogenous CM proliferation and regeneration in adult mice post-MI (Sadek and Porrello, 2020).
In a preclinical porcine model of acute MI, intracoronary infusion of recombinant human Agrin (rhAgrin) led to improved heart function and reduced scar size within 28 days of therapy. Interestingly, although previous studies in mice highlighted Agrin’s critical role on CM proliferation by inhibiting Hippo through its binding to the dystrophin-glycoprotein complex on the surface of CMs, the effects of post-MI rhAgrin therapy in pig CM proliferation were modest and insufficient to explain the robust functional and structural improvements. However, further studies determined that rhAgrin-treated pigs exhibited reduced CM apoptosis, enhanced angiogenesis, and attenuated inflammatory responses, suggesting a potential pleiotropic role for Agrin in adult mammals (Baehr et al. 2020). Likewise, knockdown of Hippo signaling in pigs through an adeno-associated virus serotype 9 (AAV-9)-mediated delivery of a short hairpin RNA (shRNA), targeting the Hippo adaptor gene Salvador, resulted in significant improvements in heart function and scar size reduction. Intriguingly, these effects were correlated with significant, albeit modest, increases in the rate of CM proliferation, which however persisted for a longer period. Moreover, some of the beneficial effects of gene therapy were likely indirect, since animals exhibited increased mitosis even in remote zone CMs that seemingly were not infected by the virus, and an increase in small blood vessel densities (Liu et al. 2021). As a final example, a high-throughput microRNA screening identified miR-199a-3p as a critical stimulator of CM proliferation and cardiac regeneration after MI in mice. Accordingly, in a porcine model of MI, gene therapy with AAV6-mediated delivery of the human miR-199a-1 pri-miRNA gene, under the control of the constitutive CMV promoter, led to transient improvements in heart function and scar size reduction, but animals succumbed to lethal sudden cardiac arrhythmias within two months post-therapy. Histological and molecular examination suggested excessive proliferation of porcine cardiomyoblasts as the underlying cause of death, presumably due to the uncontrolled expression of miR-199a-3p (Gabisonia et al. 2019). Therefore, the latter findings raise important concerns regarding the safety, efficacy, and potential oncologic risk of gene therapies, particularly for those directed towards perturbing cell cycle activity in the adult heart; and underscore the importance of carefully choosing the viral vector delivery system, dosage, and the regulatory elements driving expression of the target gene, before testing in animals or humans.
Conclusions
The transient regenerative potential seen in newborn mammals is perhaps the strongest indicator thus far that adult heart regeneration will eventually become therapeutically feasible in humans. The early premise that this would be as simple as perturbing the expression of a single gene or exogenously remuscularizing the scarred tissue with lab-grown stem cells and CMs is gradually waning, as new research findings unravel the intricacies of the path to regeneration. Epimorphic regeneration in the adult zebrafish or neonatal mouse heart requires spatiotemporal control of metabolic, EMT and growth programs that, collectively, resemble those of trabeculae corticalization or compaction during fish and mouse cardiac chamber morphogenesis, respectively. Among others, these include activation of NC-like GRNs, Nrg/ErbB- and Hif1α-mediated induction of glucose metabolism, as well as nerve-dependence, mechanotransduction and growth factor signals that regulate scar resorption, Hippo signaling and CM proliferation. Many of these factors are already being therapeutically targeted through cell- and gene-based strategies aimed at evoking adult CM regeneration from within.
Abbreviations
αMHC, alpha myosin heavy chain ; aSHF, anterior secondary heart field ; bHLH, basic helix loop helix ; DTA, diphtheria toxin A ; E, Embryonic day ; EMT, epithelial-mesenchymal transition ; CM, cardiomyocyte ; FHF, first heart field ; GMT, Gata4 Mef2c Tbx5 ; GRN, gene regulatory network ; HF, heart failure ; HLA, human leukocyte antigen ; hPSC, human pluripotent stem cells ; iPSC, induced pluripotent stem cells ; MHC, major histocompatibility complex ; MI, myocardial infarction ; MSCs, mesenchymal stem cells ; NC, neural crest ; PN, postnatal ; PS, primitive streak ; pSHF, posterior secondary heart field ; RA, retinoic acid ; RV, right ventricle ; rhAgrin, recombinant human Agrin ;References
Abdul-Wajid S., Demarest B. L., Yost H. J. (2018). Loss of embryonic neural crest derived cardiomyocytes causes adult onset hypertrophic cardiomyopathy in zebrafish. Nature Communications 9: 4603.
Aharonov A., Shakked A., Umansky K. B., Savidor A., Genzelinakh A., Kain D., Lendengolts D., Revach O.Y., Morikawa Y., Dong J., Levin Y., Geiger B., Martin J. F., Tzahor E. (2020). ERBB2 drives YAP activation and EMT-like processes during cardiac regeneration. Nature Cell Biology 22: 1346-1356.
Aurora A. B., Porrello E. R., Tan W., Mahmoud A. I., Hill J. A., Bassel-Duby R., Sadek H. A., Olson E. N. (2014). Macrophages are required for neonatal heart regeneration. Journal of Clinical Investigation 124: 1382-1392.
Bardot E., Calderon D., Santoriello F., Han S., Cheung K., Jadhav B., Burtscher I., Artap S., Jain R., Epstein J., Lickert H., Gouon-Evans V., Sharp A. J., Dubois N. C. (2017). Foxa2 identifies a cardiac progenitor population with ventricular differentiation potential. Nature Communications 8: 14428.
Bertrand N., Roux M., Ryckebüsch L., Niederreither K., Dollé P., Moon A., Capecchi M., Zaffran S. (2011). Hox genes define distinct progenitor sub-domains within the second heart field. Developmental Biology 353: 266-274.
Bise T., Sallin P., Pfefferli C., Jaźwińska A. (2020). Multiple cryoinjuries modulate the efficiency of zebrafish heart regeneration. Scientific Reports 10: 11551.
Bolli R., Mitrani R. D., Hare J. M., Pepine C. J., Perin E. C., Willerson J. T., Traverse J. H., Henry T. D., Yang P. C., Murphy M. P., March K. L., Schulman I. H., Ikram S., Lee D. P., O'Brien C., Lima J. A., Ostovaneh M. R., Ambale‐Venkatesh B., Lewis G., Khan A., Bacallao K., Valasaki K., Longsomboon B., Gee A. P., Richman S., Taylor D. A., Lai D., Sayre S. L., Bettencourt J., Vojvodic R. W., Cohen M. L., Simpson L., Aguilar D., Loghin C., Moyé L., Ebert R. F., Davis B. R., Simari R. D. (2021). A Phase II study of autologous mesenchymal stromal cells and c‐kit positive cardiac cells, alone or in combination, in patients with ischaemic heart failure: the CCTRN CONCERT‐HF trial . European Journal of Heart Failure 23: 661-674.
Cai M., Shen R., Song L., Lu M., Wang J., Zhao S., Tang Y., Meng X., Li Z., He Z.X. (2016). Bone Marrow Mesenchymal Stem Cells (BM-MSCs) Improve Heart Function in Swine Myocardial Infarction Model through Paracrine Effects. Scientific Reports 6: 28250.
Chabab S., Lescroart F., Rulands S., Mathiah N., Simons B. D., Blanpain C. (2016). Uncovering the Number and Clonal Dynamics of Mesp1 Progenitors during Heart Morphogenesis. Cell Reports 14: 1-10.
Christiaen L., Stolfi A., Davidson B., Levine M., (2009). Spatio-temporal intersection of Lhx3 and Tbx6 defines the cardiac field through synergistic activation of Mesp. Developmental Biology 328: 552-560.
Costello I., Pimeisl I.M., Dräger S., Bikoff E. K., Robertson E. J., Arnold S. J. (2011). The T-box transcription factor Eomesodermin acts upstream of Mesp1 to specify cardiac mesoderm during mouse gastrulation. Nature Cell Biology 13: 1084-1091.
Das S., Goldstone A. B., Wang H., Farry J., D’Amato G., Paulsen M. J., Eskandari A., Hironaka C. E., Phansalkar R., Sharma B., Rhee S., Shamskhou E. A., Agalliu D., de Jesus Perez V., Woo Y. J., Red-Horse K. (2019). A Unique Collateral Artery Development Program Promotes Neonatal Heart Regeneration. Cell 176: 1128-1142.e18.
David R., Brenner C., Stieber J., Schwarz F., Brunner S., Vollmer M., Mentele E., Müller-Höcker J., Kitajima S., Lickert H., Rupp R., Franz W.M. (2008). MesP1 drives vertebrate cardiovascular differentiation through Dkk-1-mediated blockade of Wnt-signalling. Nature Cell Biology 10: 338-345.
Dehal P., Satou Y., Campbell R. K., Chapman J., Degnan B., De Tomaso A., Davidson B., Di Gregorio A., Gelpke M., Goodstein D. M., Harafuji N., Hastings K. E. M., Ho I., Hotta K., Huang W., Kawashima T., Lemaire P., Martinez D., Meinertzhagen I. A., Necula S., Nonaka M., Putnam N., Rash S., Saiga H., Satake M., Terry A., Yamada L., Wang H.G., Awazu S., Azumi K., Boore J., Branno M., Chin-bow S., DeSantis R., Doyle S., Francino P., Keys D. N., Haga S., Hayashi H., Hino K., Imai K. S., Inaba K., Kano S., Kobayashi K., Kobayashi M., Lee B.I., Makabe K. W., Manohar C., Matassi G., Medina M., Mochizuki Y., Mount S., Morishita T., Miura S., Nakayama A., Nishizaka S., Nomoto H., Ohta F., Oishi K., Rigoutsos I., Sano M., Sasaki A., Sasakura Y., Shoguchi E., Shin-i T., Spagnuolo A., Stainier D., Suzuki M. M., Tassy O., Takatori N., Tokuoka M., Yagi K., Yoshizaki F., Wada S., Zhang C., Hyatt P. D., Larimer F., Detter C., Doggett N., Glavina T., Hawkins T., Richardson P., Lucas S., Kohara Y., Levine M., Satoh N., Rokhsar D. S. (2002). The Draft Genome of Ciona intestinalis : Insights into Chordate and Vertebrate Origins . Science 298: 2157-2167.
del Monte-Nieto G., Ramialison M., Adam A. A. S., Wu B., Aharonov A., D’Uva G., Bourke L. M., Pitulescu M. E., Chen H., de la Pompa J. L., Shou W., Adams R. H., Harten S. K., Tzahor E., Zhou B., Harvey R. P. (2018). Control of cardiac jelly dynamics by NOTCH1 and NRG1 defines the building plan for trabeculation. Nature 557: 439-445.
Ehrman L. A., Yutzey K. E. (1999). Lack of Regulation in the Heart Forming Region of Avian Embryos. Developmental Biology 207: 163-175.
Elhelaly W. M., Cardoso A. C., Pereira A. H. M., Elnawasany A., Ebrahimi S., Nakada Y., Sadek H. A. (2019). C-Kit Cells Do Not Significantly Contribute to Cardiomyogenesis During Neonatal Heart Regeneration. Circulation 139: 559-561.
Eschenhagen T., Bolli R., Braun T., Field L. J., Fleischmann B. K., Frisén J., Giacca M., Hare J. M., Houser S., Lee R. T., Marbán E., Martin J. F., Molkentin J. D., Murry C. E., Riley P. R., Ruiz-Lozano P., Sadek H. A., Sussman M. A., Hill J. A. (2017). Cardiomyocyte Regeneration. Circulation 136: 680-686.
Farkas J. E., Monaghan J. R. (2017). A brief history of the study of nerve dependent regeneration. Neurogenesis 4: e1302216.
Firulli A. B., McFadden D. G., Lin Q., Srivastava D., Olson E. N. (1998). Heart and extra-embryonic mesodermal defects in mouse embryos lacking the bHLH transcription factor Hand1. Nature Genetics 18: 266-270.
Fukuda R., Aharonov A., Ong Y. T., Stone O. A., El-Brolosy M., Maischein H.M., Potente M., Tzahor E., Stainier D. Y.R. (2019). Metabolic modulation regulates cardiac wall morphogenesis in zebrafish. eLife 8: e50161.
Guimarães-Camboa N., Stowe J., Aneas I., Sakabe N., Cattaneo P., Henderson L., Kilberg M. S., Johnson R. S., Chen J., McCulloch A. D., Nobrega M. A., Evans S. M., Zambon A. C. (2015). HIF1α Represses Cell Stress Pathways to Allow Proliferation of Hypoxic Fetal Cardiomyocytes. Developmental Cell 33: 507-521.
Gupta V., Poss K. D. (2012). Clonally dominant cardiomyocytes direct heart morphogenesis. Nature 484: 479-484.
Harvey R. P., Wystub-Lis K., del Monte-Nieto G., Graham R. M., Tzahor E. (2016). Cardiac Regeneration Therapies – Targeting Neuregulin 1 Signalling. Heart, Lung and Circulation 25: 4-7.
Hashimoto H., Olson E. N., Bassel-Duby R. (2018). Therapeutic approaches for cardiac regeneration and repair. Nature Reviews Cardiology 15: 585-600.
Hatzistergos K. E., Durante M. A., Valasaki K., Wanschel A. C. B. A., Harbour J. W., Hare J. M. (2020). A novel cardiomyogenic role for Isl1 + neural crest cells in the inflow tract . Science Advances 6: eaba9950.
Hatzistergos K. E., Quevedo H., Oskouei B. N., Hu Q., Feigenbaum G. S., Margitich I. S., Mazhari R., Boyle A. J., Zambrano J. P., Rodriguez J. E., Dulce R., Pattany P. M., Valdes D., Revilla C., Heldman A. W., McNiece I., Hare J. M. (2010). Bone Marrow Mesenchymal Stem Cells Stimulate Cardiac Stem Cell Proliferation and Differentiation. Circulation Research 107: 913-922.
Hatzistergos K. E., Saur D., Seidler B., Balkan W., Breton M., Valasaki K., Takeuchi L. M., Landin A. M., Khan A., Hare J. M. (2016). Stimulatory Effects of Mesenchymal Stem Cells on cKit + Cardiac Stem Cells Are Mediated by SDF1/CXCR4 and SCF/cKit Signaling Pathways . Circulation Research 119: 921-930.
Hatzistergos K. E., Takeuchi L. M., Saur D., Seidler B., Dymecki S. M., Mai J. J., White I. A., Balkan W., Kanashiro-Takeuchi R. M., Schally A. V., Hare J. M. (2015). cKit + cardiac progenitors of neural crest origin . Proceedings of the National Academy of Sciences 112: 13051-13056.
Honkoop H., de Bakker D. E.M., Aharonov A., Kruse F., Shakked A., Nguyen P. D., de Heus C., Garric L., Muraro M. J., Shoffner A., Tessadori F., Peterson J. C., Noort W., Bertozzi A., Weidinger G., Posthuma G., Grün D., van der Laarse W. J., Klumperman J., Jaspers R. T., Poss K. D., van Oudenaarden A., Tzahor E., Bakkers J. (2019). Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. eLife 8: e50163.
Icardo J. M., Fernandez-Terán A. (1987). Morphologic Study of Ventricular Trabeculation in the Embryonic Chick Heart. Cells Tissues Organs 130: 264-274.
Jain R., Li D., Gupta M., Manderfield L. J., Ifkovits J. L., Wang Q., Liu F., Liu Y., Poleshko A., Padmanabhan A., Raum J. C., Li L., Morrisey E. E., Lu M. M., Won K.J., Epstein J. A. (2015). Integration of Bmp and Wnt signaling by Hopx specifies commitment of cardiomyoblasts. Science 348: aaa6071.
Jesty S. A., Steffey M. A., Lee F. K., Breitbach M., Hesse M., Reining S., Lee J. C., Doran R. M., Nikitin A. Y., Fleischmann B. K., Kotlikoff M. I. (2012). c-kit+ precursors support postinfarction myogenesis in the neonatal, but not adult, heart. Proceedings of the National Academy of Sciences 109: 13380-13385.
Konfino T., Landa N., Ben‐Mordechai T., Leor J. (2015). The Type of Injury Dictates the Mode of Repair in Neonatal and Adult Heart. Journal of the American Heart Association 4: e001320.
Konishi A., Sakushima K., Isobe S., Sato D. (2016). First Approval of Regenerative Medical Products under the PMD Act in Japan. Cell Stem Cell 18: 434-435.
Kumar A., Godwin J. W., Gates P. B., Garza-Garcia A. A., Brockes J. P. (2007). Molecular Basis for the Nerve Dependence of Limb Regeneration in an Adult Vertebrate. Science 318: 772-777.
Lee J. H., Protze S. I., Laksman Z., Backx P. H., Keller G. M. (2017). Human Pluripotent Stem Cell-Derived Atrial and Ventricular Cardiomyocytes Develop from Distinct Mesoderm Populations. Cell Stem Cell 21: 179-194.e4.
Lee Y. M., Jeong C.H., Koo S.Y., Son M. J., Song H. S., Bae S.K., Raleigh J. A., Chung H.Y., Yoo M.A., Kim K.W. (2001). Determination of hypoxic region by hypoxia marker in developing mouse embryos in vivo: A possible signal for vessel development. Developmental Dynamics 220: 175-186.
Li Y., Lv Z., He L., Huang X., Zhang S., Zhao H., Pu W., Li Y., Yu W., Zhang L., Liu X., Liu K., Tang J., Tian X., Wang Q.D., Lui K. O., Zhou B. (2019). Genetic Tracing Identifies Early Segregation of the Cardiomyocyte and Nonmyocyte Lineages. Circulation Research 125: 343-355.
Liu X., De la Cruz E., Gu X., Balint L., Oxendine-Burns M., Terrones T., Ma W., Kuo H.H., Lantz C., Bansal T., Thorp E., Burridge P., Jakus Z., Herz J., Cleaver O., Torres M., Oliver G. (2020). Lymphoangiocrine signals promote cardiac growth and repair. Nature 588: 705-711.
Luo W., Hu H., Chang R., Zhong J., Knabel M., O'Meally R., Cole R. N., Pandey A., Semenza G. L. (2011). Pyruvate Kinase M2 Is a PHD3-Stimulated Coactivator for Hypoxia-Inducible Factor 1. Cell 145: 732-744.
Mahmoud A. I., O’Meara C. C., Gemberling M., Zhao L., Bryant D. M., Zheng R., Gannon J. B., Cai L., Choi W.Y., Egnaczyk G. F., Burns C. E., Burns C. G., MacRae C. A., Poss K. D., Lee R. T. (2015). Nerves Regulate Cardiomyocyte Proliferation and Heart Regeneration. Developmental Cell 34: 387-399.
Menasché P. (2018). Cell therapy trials for heart regeneration — lessons learned and future directions. Nature Reviews Cardiology 15: 659-671.
Menendez-Montes I., Escobar B., Palacios B., Gómez M. J., Izquierdo-Garcia J. L., Flores L., Jiménez-Borreguero L. J., Aragones J., Ruiz-Cabello J., Torres M., Martin-Puig S. (2016). Myocardial VHL-HIF Signaling Controls an Embryonic Metabolic Switch Essential for Cardiac Maturation. Developmental Cell 39: 724-739.
Nguyen P. D., de Bakker D. E. M., Bakkers J. (2021). Cardiac regenerative capacity: an evolutionary afterthought?. Cellular and Molecular Life Sciences 78: 5107-5122.
Park S.J., Kim R. Y., Park B.W., Lee S., Choi S. W., Park J.H., Choi J. J., Kim S.W., Jang J., Cho D.W., Chung H.M., Moon S.H., Ban K., Park H.J. (2019). Dual stem cell therapy synergistically improves cardiac function and vascular regeneration following myocardial infarction. Nature Communications 10: 3123.
Pashmforoush M., Lu J. T., Chen H., Amand T. S., Kondo R., Pradervand S., Evans S. M., Clark B., Feramisco J. R., Giles W., Ho S. Y., Benson D.W., Silberbach M., Shou W., Chien K. R. (2004). Nkx2-5 Pathways and Congenital Heart Disease. Cell 117: 373-386.
Porrello E. R., Mahmoud A. I., Simpson E., Hill J. A., Richardson J. A., Olson E. N., Sadek H. A. (2011). Transient Regenerative Potential of the Neonatal Mouse Heart. Science 331: 1078-1080.
Poss K. D., Wilson L. G., Keating M. T. (2002). Heart Regeneration in Zebrafish. Science 298: 2188-2190.
Prall O. W.J., Menon M. K., Solloway M. J., Watanabe Y., Zaffran S., Bajolle F., Biben C., McBride J. J., Robertson B. R., Chaulet H., Stennard F. A., Wise N., Schaft D., Wolstein O., Furtado M. B., Shiratori H., Chien K. R., Hamada H., Black B. L., Saga Y., Robertson E. J., Buckingham M. E., Harvey R. P. (2007). An Nkx2-5/Bmp2/Smad1 Negative Feedback Loop Controls Heart Progenitor Specification and Proliferation. Cell 128: 947-959.
Priya R., Allanki S., Gentile A., Mansingh S., Uribe V., Maischein H.M., Stainier D. Y. R. (2020). Tension heterogeneity directs form and fate to pattern the myocardial wall. Nature 588: 130-134.
Qin X., Lin L., Cao L., Zhang X., Song X., Hao J., Zhang Y., Wei R., Huang X., Lu J., Ge Q. (2017). Extracellular matrix protein Reelin promotes myeloma progression by facilitating tumor cell proliferation and glycolysis. Scientific Reports 7: 45305.
Ream M., Ray A. M., Chandra R., Chikaraishi D. M. (2008). Early fetal hypoxia leads to growth restriction and myocardial thinning. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology 295: R583-R595.
Sadek H. A., Porrello E. R. (2020). Neonatal heart regeneration: Moving from phenomenology to regenerative medicine. The Journal of Thoracic and Cardiovascular Surgery 159: 2451-2455.
Sallin P., de Preux Charles A.S., Duruz V., Pfefferli C., Jaźwińska A. (2015). A dual epimorphic and compensatory mode of heart regeneration in zebrafish. Developmental Biology 399: 27-40.
Sánchez-Iranzo H., Galardi-Castilla M., Minguillón C., Sanz-Morejón A., González-Rosa J. M., Felker A., Ernst A., Guzmán-Martínez G., Mosimann C., Mercader N. (2018). Tbx5a lineage tracing shows cardiomyocyte plasticity during zebrafish heart regeneration. Nature Communications 9: 428.
Sande-Melón M., Marques I. J., Galardi-Castilla M., Langa X., Pérez-López M., Botos M.A., Sánchez-Iranzo H., Guzmán-Martínez G., Ferreira Francisco D. M., Pavlinic D., Benes V., Bruggmann R., Mercader N. (2019). Adult sox10+ Cardiomyocytes Contribute to Myocardial Regeneration in the Zebrafish. Cell Reports 29: 1041-1054.e5.
Shiba Y., Gomibuchi T., Seto T., Wada Y., Ichimura H., Tanaka Y., Ogasawara T., Okada K., Shiba N., Sakamoto K., Ido D., Shiina T., Ohkura M., Nakai J., Uno N., Kazuki Y., Oshimura M., Minami I., Ikeda U. (2016). Allogeneic transplantation of iPS cell-derived cardiomyocytes regenerates primate hearts. Nature 538: 388-391.
Tang W., Martik M. L., Li Y., Bronner M. E. (2019). Cardiac neural crest contributes to cardiomyocytes in amniotes and heart regeneration in zebrafish. eLife 8: e47929.
Tonegawa A., Takahashi Y. (1998). Somitogenesis Controlled by Noggin. Developmental Biology 202: 172-182.
Tyser R. C. V., Ibarra-Soria X., McDole K., Arcot Jayaram S., Godwin J., van den Brand T. A. H., Miranda A. M. A., Scialdone A., Keller P. J., Marioni J. C., Srinivas S. (2021). Characterization of a common progenitor pool of the epicardium and myocardium. Science 371: eabb2986.
Vagnozzi R. J., Maillet M., Sargent M. A., Khalil H., Johansen A. K. Z., Schwanekamp J. A., York A. J., Huang V., Nahrendorf M., Sadayappan S., Molkentin J. D. (2020). An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 577: 405-409.
Wang J., Liu S., Heallen T., Martin J. F. (2018). The Hippo pathway in the heart: pivotal roles in development, disease, and regeneration. Nature Reviews Cardiology 15: 672-684.
Wang J., Panáková D., Kikuchi K., Holdway J. E., Gemberling M., Burris J. S., Singh S. P., Dickson A. L., Lin Y.F., Sabeh M. K., Werdich A. A., Yelon D., MacRae C. A., Poss K. D. (2011). The regenerative capacity of zebrafish reverses cardiac failure caused by genetic cardiomyocyte depletion. Development 138: 3421-3430.
White I. A., Gordon J., Balkan W., Hare J. M. (2015). Sympathetic Reinnervation Is Required for Mammalian Cardiac Regeneration. Circulation Research 117: 990-994.
Xu H., Wang B., Ono M., Kagita A., Fujii K., Sasakawa N., Ueda T., Gee P., Nishikawa M., Nomura M., Kitaoka F., Takahashi T., Okita K., Yoshida Y., Kaneko S., Hotta A. (2019). Targeted Disruption of HLA Genes via CRISPR-Cas9 Generates iPSCs with Enhanced Immune Compatibility. Cell Stem Cell 24: 566-578.e7.
Yamanaka S. (2020). Pluripotent Stem Cell-Based Cell Therapy—Promise and Challenges. Cell Stem Cell 27: 523-531.
Yuan X., Qi H., Li X., Wu F., Fang J., Bober E., Dobreva G., Zhou Y., Braun T. (2017). Disruption of spatiotemporal hypoxic signaling causes congenital heart disease in mice. Journal of Clinical Investigation 127: 2235-2248.
Yue Y., Zong W., Li X., Li J., Zhang Y., Wu R., Liu Y., Cui J., Wang Q., Bian Y., Yu X., Liu Y., Tan G., Zhang Y., Zhao G., Zhou B., Chen L., Xiao W., Cheng H., He A. (2020). Long-term, in toto live imaging of cardiomyocyte behaviour during mouse ventricle chamber formation at single-cell resolution. Nature Cell Biology 22: 332-340.
Zhang Q., Carlin D., Zhu F., Cattaneo P., Ideker T., Evans S. M., Bloomekatz J., Chi N. C. (2021). Unveiling Complexity and Multipotentiality of Early Heart Fields. Circulation Research 129: 474-487.
Zhao M., Nakada Y., Wei Y., Bian W., Chu Y., Borovjagin A. V., Xie M., Zhu W., Nguyen T., Zhou Y., Serpooshan V., Walcott G. P., Zhang J. (2021). Cyclin D2 Overexpression Enhances the Efficacy of Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes for Myocardial Repair in a Swine Model of Myocardial Infarction. Circulation 144: 210-228.