Int. J. Dev. Biol. 68: 135 - 143 (2024)
Fibroblast Growth Factor 8 enhances the chondrogenesis of trunk neural crest cells: a possible gene regulatory network
Open Access | Original Article | Published: 12 December 2024
Abstract
The neural crest (NC) is an embryonic cell population with high migratory capacity. It contributes to forming several organs and tissues, such as the craniofacial skeleton and the peripheral nervous system of vertebrates. Both pre-migratory and post-migratory NC cells are plastic, adopting multiple differentiation paths by responding to different inductive environmental signals. Cephalic neural crest cells (CNCCs) give rise to most of the cartilage and bone tissues in the head. On the other hand, the mesenchymal potential of trunk neural crest cells (TNCCs) is sparsely detected in some animal groups. The mesenchymal potential of TNCCs can be unveiled through specific environmental conditions of NC cultures. In this study, we present evidence that FGF8 treatment can foster increased chondrogenic differentiation of TNCCs, particularly during treatment at the migratory stage. Additionally, we conducted a transcriptomic analysis of TNCCs in the post-migratory stage, noting that exogenous FGF8 signaling can sustain multipotent status and, possibly, at the same time, a pro-cartilage regulatory gene network. Our results provide a more comprehensive understanding of the mechanisms underlying chondrogenic differentiation from TNCCs.
Keywords
trunk neural crest cells, FGF8, cartilage, multipotency, skeletogenic potential, transcriptome
Introduction
The neural crest (NC) is a transient, multipotent population of cells that arises during early vertebrate development at the border of the developing nervous system (Le Douarin and Kalcheim, 1999). After their specification and delamination, the NC cells (NCCs) migrate through defined routes to almost all regions of the developing embryo body. Once at their final destination, NCCs contribute to distinct cell types such as neurons, glial cells, melanocytes, adipocytes, chondrocytes, and osteocytes (Prasad et al., 2019).
For many years, it was considered that only NCCs from the cranial region (CNCCs) were capable of originating cartilage and bone, forming the most extensive parts of the craniofacial skeleton of vertebrates (Nakamura and Ayer-le Lievre, 1982; Noden, 1978). However, over the years, several studies suggest that TNCCs could also contribute to distinct skeletogenic elements, including the lepidothichia of fishes, some parts of the turtle’s shell, and the dermal plates of armadillos (Gilbert et al., 2007; Kague et al., 2012; Krmpotic et al., 2021). Although disputed, evidence for the skeletogenic potential of TNCCs has been provided by in vitro experiments showing cartilage and bone differentiation of avian and mouse TNCCs (for an in-depth review on the subject, see Rodrigues-da-Silva et al., 2022).
The chondrogenic potential exhibited by avian and mouse TNCCs in vitro raises questions about why this potential is not expressed in vivo in such amniotes. Is it due to the absence of necessary signals or the presence of inhibitory signals? Furthermore, are TNCCs already committed to neural and melanocytic fates preventing chondrogenic potential expression? If so, is this restriction imposed by a gene regulatory network (GRN)? In the affirmative case, can this GRN be regulated by extracellular stimuli to allow for the expression of chondrogenic potential? Understanding such mechanisms could provide valuable evolutionary clues about why TNCCs express their mesenchymal potential in vivo at certain times and not others. In vitro studies can help us find answers to these questions.
Different environmental conditions can unveil the skeletogenic potential of TNCCs, such as the time of cell culture (McGonnell and Graham, 2002; Abzhanov, 2003) and the use of specific substrates. In the last case, MatrigelTM and PuramatrixTM significantly enhanced the frequency and number of cartilage nodules compared to cultures performed under fibroblast feeder layers (Ramos-Hryb et al., 2013; Taufer et al., 2020).
Moreover, signaling molecules can also stimulate chondrogenesis from TNCCs, like the morphogen Sonic Hedgehog (SHH) (Calloni et al., 2007; Calloni et al., 2009) and FGF2, a member of the fibroblast growth factor family (Ido and Ito 2006). At the cephalic level, we found another member of the FGF family, FGF8, which is secreted by the isthmic brain-signaling center during NCC’s delamination. FGF8 alters the expression of HOXA2 and affects the patterns of bone formation of CNCCs (Trainor et al., 2002). More recently, our research group showed that FGF8 and SHH added together can strongly stimulate chondrogenesis in cultures of mesencephalic NCCs (MNCCs) (da Costa et al., 2018). However, higher rates of chondrogenesis were obtained when MNCCs were treated with FGF8 during the migration phase, thus mimicking the in vivo exposure of migrating NCCs to FGF8 secreted by the isthmus. Although there is no signaling center, such as the isthmus releasing FGF8 in the trunk region of vertebrates, we wondered about the possible effects of treating TNCCs with FGF8 on chondrogenesis.
Here, we show that FGF8 added to TNCCs at the migration phase can increase cartilage nodules' number and frequency. This effect was correlated to the increase in the expression of some genes necessary for the chondrogenic program to take place, revealing that FGF8 can stimulate chondrogenesis not only at cephalic but also at truncal levels. Based on the transcriptional data, we propose two main scenarios that could help explain how FGF8 stimulates TNCCs chondrogenesis.
Results
FGF8 promotes chondrogenesis by TNCCs in culture
Several works have already demonstrated that FGF8 is crucial for cranial NCCs (CNCCs) chondrogenesis both in vivo and in vitro (Abzhanov et al., 2003; Abzhanov et al.,2007; Abzhanov and Tabin, 2004; Creuzet et al., 2004; Hackland et al., 2019; Shao et al., 2015). Previously, our group demonstrated that adding both FGF8 and SHH during 48 h of secondary culture strongly promotes chondrogenesis and the multipotentiality of mesencephalic NCCs progenitors (MNCCs) (da Costa et al., 2018). More importantly, FGF8 added to migrating MNCCs during the primary cultures (15 hours) can potentiate this chondrogenic effect on NC (da Costa et al., 2018).
Here, we analyze the role of FGF8 at different doses and exposure times in stimulating chondrogenesis in TNCCs. The primary cultures of TNCCs correspond to the first 15 hours of cell migration, and FGF8 was administered at a concentration of 100 ng/ml. Secondary cultures correspond to migrated TNCCs harvested and replated for an increasing period of 10 days of culture. In the last case, FGF8 was administered at two different concentrations (10 and 100 ng/ml). After ten days of culture, cartilage nodules were identified using brightfield microscopy (Fig. 1A) and immunocytochemical staining with a chondroitin sulfate antibody (Fig. 1B).
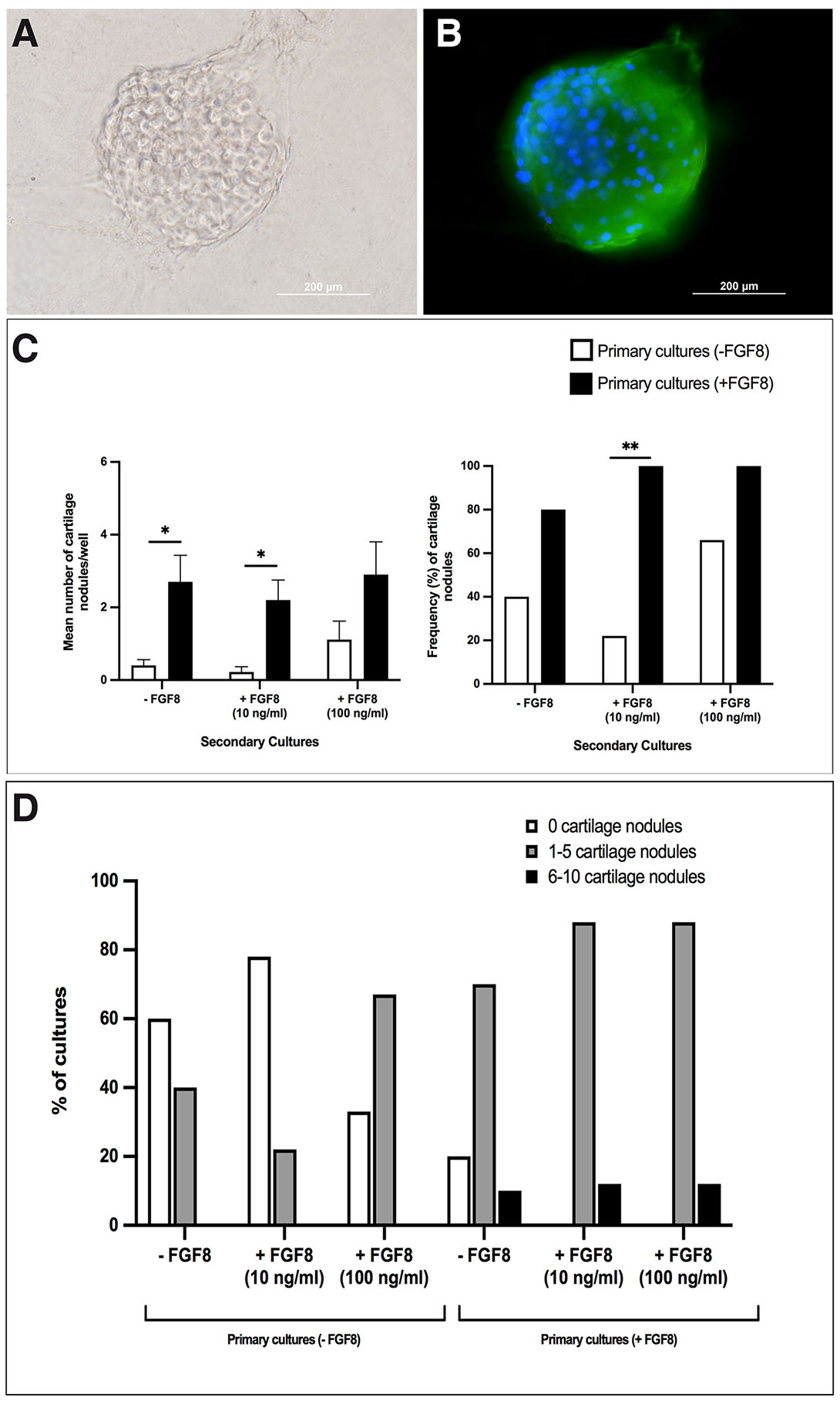
Fig. 1. FGF8 treatment enhances chondrogenic differentiation in trunk neural crest (TNC) cultures.
Representative images of cartilage nodules evidenced by bright field microscopy (A) and chondroitin sulfate (CS) immunofluorescence (B); green, chondroitin sulfate; blue, DAPI. Scale bar, 200 µm. (C) Graphs quantifying the number of cartilage nodules (left) and the frequency of wells (right) containing cartilage nodules. (D) Graph quantifying the frequency of cultures according to increasing numbers of cartilage nodules. TNC cultures were analyzed after ten days of secondary cultures from TNCCs treated (10 and 100 ng/ml) or not with FGF8 during the migratory stage. Graph on the left in (C): the analysis was performed using Student’s t-test. Results were expressed as Mean + SEM and considered significant where *P<0.01. Graph on the right in (C): the analysis was performed by X2 test where *P<0.01; **P<0.001. Data was obtained from three independent experiments (n=10 for each condition).
Cartilage nodules were detected in all secondary TNCC cultures performed on Matrigel™ after ten days. However, FGF8 treatment increased cartilage nodules' number and frequency (Fig. 1C). For example, adding FGF8 only during the 15 hours of the primary cultures promoted around 7-fold increase (p=0.006) in the number of cartilage nodules compared to FGF8-untreated cultures (Fig. 1C – left graph). Cultures that received FGF8 (100 ng/mL) during the primary cultures and were later treated with 10 ng/mL FGF8 in secondary cultures displayed a 12-fold increase (p=0.002) in the number of cartilage nodules compared to TNC cultures that were not fed by FGF8 during the migration period.
Finally, TNCC cultures treated with FGF8 100 ng/ml continuously (i.e., at both primary and secondary cultures) presented the highest rates of chondrogenesis, with an average of three cartilage nodules per well. However, TNCC cultures treated with 100 ng/ml FGF8 only at secondary cultures also displayed elevated rates of chondrogenesis. It explains why this data was not statistically significant despite a 3-fold increase in cartilage nodules in cultures treated continuously with FGF8 (p=0.06).
As mentioned, FGF8 treatment during the migratory stage also increased the frequency of the appearance of cartilage nodules in TNCC cultures (Fig. 1C – right graph). For example, in cultures that did not receive FGF8 at any moment, the frequency of appearance of cartilage nodules was 40%. In contrast, in cultures that received FGF8 only during the migratory stage, this percentage doubled to 80%, p=0.067. Cultures fed with FGF8 during the migratory phase and later with FGF8 at 10 ng/ml or 100 ng/ml during secondary cultures displayed 100% of culture wells with cartilage nodules. On the other hand, cartilage nodules were detected in only 22% of the wells (p= 0.0007) that received 10 ng/ml FGF8 only at the secondary cultures. Finally, in cultures treated with 100 ng/ml of FGF8 only during the secondary cultures, 66% of the wells presented cartilage nodules, p=0.057.
The analysis of cartilage nodules distribution in function of FGF8 treatment (Fig. 1D) indicates that all secondary cultures in which primary cultures were treated with FGF8 exhibited the highest number of cartilage nodules, ranging from 6 to 10. Even in secondary cultures that did not receive FGF8, approximately 10% of the culture wells displayed 6 to 10 cartilage nodules, similar to secondary cultures treated with 10 or 100 ng/ml of FGF8. This data highlights the importance of FGF8 added to primary cultures to stimulate the chondrogenesis observed in secondary cultures.
Therefore, the treatment with FGF8 during the 15 hours of cell migration significantly increased the number and frequency of cartilage nodules in secondary cultures compared to TNCC cultures not treated with FGF8 during the migratory phase.
FGF8 enhances the expression of stemness and cartilage-related genes by TNCCs in culture
Based on these observations, we next investigated the transcriptional profile of TNCCs submitted to FGF8 treatment. Therefore, we performed RNA-seq analysis of TNCCs at the end of the primary cultures at 15 hours of cell migration. Distance analysis between samples, using hierarchical clustering (heatmap) and principal component analysis (PCA), revealed significant differences in the transcriptional profiles of samples treated with FGF8 (+FGF8) compared to untreated samples (-FGF8). This differential expression pattern, visualized through the heatmap and PCA, indicates specific transcriptional changes induced by FGF8 (Supplementary Figs. 1 and 2). A total of 9164 transcripts were identified, of which 511 were upregulated and 460 downregulated under FGF8 treatment (Fig. 2A and Supplementary Table 1).
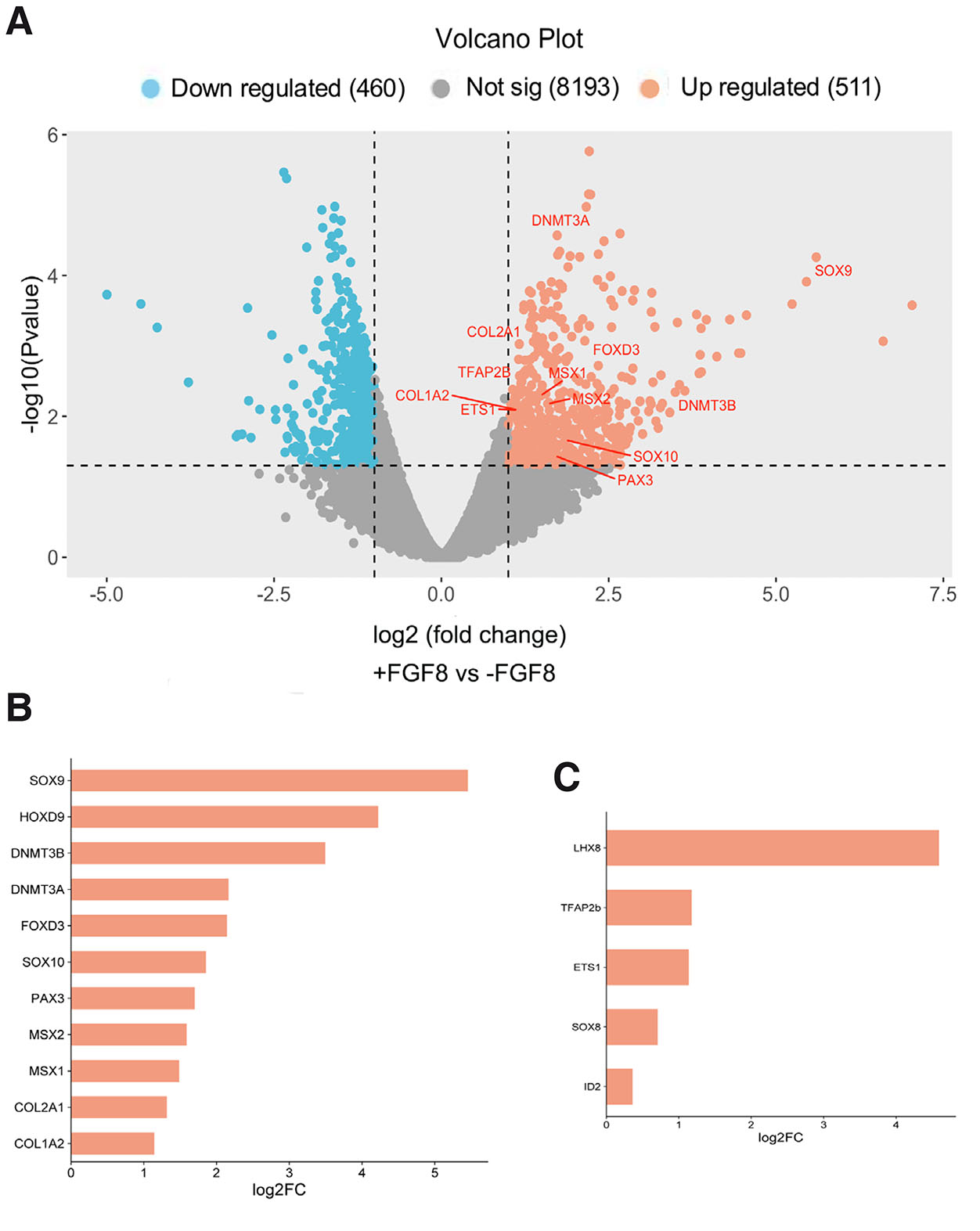
Fig. 2. Expression profile of key genes following FGF8 treatment.
(A) The volcano plot illustrates differentially expressed genes in migratory trunk neural crest cells (TNCCs) after 15 hours of FGF8 treatment. The x-axis represents the log2-fold change in gene expression, and the y-axis shows the -log10 p-value, indicating the statistical significance of each gene. Upregulated genes are highlighted in red, while downregulated genes are depicted in blue. Enrichment levels of genes related to neural crest specification, multipotency, epigenetics, chondrogenesis (B), and cranial identity (C) expressed at 15 hours in migratory TNCCs treated with FGF8.
TNCCs treated with FGF8 revealed an expression profile closer to cells in earlier stages of development, featuring bona fide specification modules, such as canonical NC transcriptional factors SOX10, FOXD3, MSX1, MSX2, and PAX3 (Fig. 2A). All these genes showed at least a one-fold increase compared to cells not treated with FGF8 (Fig. 2B). Moreover, some genes related to cartilage appearance (HOXD9, SOX9, COL2A1) were upregulated under FGF8 treatment. Notably, SOX9 - a gene critical for chondrogenic differentiation - was upregulated 44-fold upon FGF8 treatment. Additionally, the upregulation of two key genes involved in epigenetic modifications (DNMT3A and DNMT3B) suggests that FGF8 may affect cartilage formation in TNCCs through epigenetic mechanisms. Finally, some genes with cranial identity were detected in TNCCs like LHX8, TFAP2b, ETS1, SOX8, and ID2 (Fig. 2C). HOXD9 and LHX8 were excluded from the differential expression analysis due to not meeting the CPM (Counts Per Million) > 1 filter. However, they showed counts exclusively in the treated group in the normalized reads using TMM (Trimmed Mean of M-values). Therefore, the expression of HOXD9 and LHX8 exclusively in FGF8-treated TNC cultures indicates that the growth factor may regulate these crucial genes related to mesenchymal lineages.
Discussion
The first important point is that our data have shown that chondrogenesis from TNCCs can occur without FGF8. However, adding FGF8 can significantly increase the number of cartilage nodules produced. Therefore, TNCCs are endowed with a chondrogenic potential that can be unveiled under certain environmental conditions, and FGF8 amplifies this potential rather than activating it or making it possible.
A second important point to highlight is that TNCC cultures treated with FGF8 only during the first 15 hours have exhibited similar levels of chondrogenesis as TNC cultures that received FGF8 throughout primary and secondary cultures. Therefore, FGF8 can stimulate chondrogenesis from the beginning of TNCC's development during the pre-migratory and migratory phases. Hence, based on those functional data and on the differential expression of genes obtained from the transcriptome of TNCCs treated or without FGF8 during 15 hours of migration, we will explore two possible scenarios (or hypotheses) by which FGF8 could be acting to amplify the chondrogenic potential of TNCCs. Rather than viewing these scenarios separately, they can complement and enhance each other.
FGF8 sustains TNCCs in an undifferentiated/multipotent state
Maintenance of the stem cell state involves at least three distinct functions: (1) inhibition of overt differentiation, (2) maintenance of proliferative capacity, and (3) maintenance of multipotency (Kim et al., 2003). We propose that FGF8 keeps TNC progenitors undifferentiated and multipotent, supporting the expansion of a neural crest cell (NCC) cluster. Maintaining this multipotent state is essential until the cells can be placed in an environment that promotes chondrogenic differentiation, such as the Matrigel substrate.
A previous study indicates that FGF8 signaling is essential for maintaining the progenitor status and multipotency of cranial neural crest cells (CNCCs) in head development (Shao et al., 2015). In mice embryos with targeted overexpression of FGF8 (Wnt1-cre; Rosa26R-FGF8), there was a complete lack of identifiable tissues or organ structures, including the tongue, bone, cartilage, and muscle. Markers for bone and cartilage differentiation, such as RUNX2 and COL2A1, were absent, as were markers for neural and myogenic differentiation, like β-TUBIII and MYOD. The migration and proliferation of neural crest cells were unaffected, leading the authors to propose that FGF8 maintains these cells in an undifferentiated state (Shao et al., 2015).
Searching for possible candidates for this effect, the authors identified MSX1, a transcription factor expressed in pre-migratory and migrating neural crest cells (NCCs), as a potential candidate in their study. In Wnt1-cre; Rosa26R-FGF8 mice, MSX1 was activated in the mandibular and part of the maxillary mesenchyme, suggesting these cells retain a progenitor status (Shao et al., 2015). MSX1 responds strongly to FGF8 overexpression in Xenopus, and FGF8 morpholino (FGF8-MO) inhibits its expression (Monsoro-Burq et al., 2005). Our FGF8 treatment showed a 2.8-fold increase in MSX1 expression, indicating its potential role in maintaining the undifferentiated and multipotent state of treated NCCs.
The other candidate is SOX9, an early transcription factor associated with NCCs, essential for maintaining the undifferentiated state of multipotent progenitor cells. In chicken embryos, high SOX9 expression in the neural tube induces a migratory NC-like phenotype and maintains these cells undifferentiated (McKeown et al., 2005). This expression is preserved in various multipotent NC-derived progenitor cells, including those from rat periodontal ligament (Techawattanawisal et al., 2007), dental sources (Degistirici et al., 2008), and skin (Zhao et al., 2009). Additionally, in Wnt1-cre; Rosa26R-FGF8 mesenchymal cells, SOX9 expression significantly increases, emphasizing its role in maintaining progenitor status under FGF8 overexpression in cranial NC cells (Shao et al., 2015).
SOX9 is essential for distinguishing NCCs from the neuroepithelium and maintaining their multipotency (Cheung and Briscoe, 2003). It guides differentiation decisions, steering NCCs towards a chondrogenic lineage rather than neuronal fates (Akiyama et al., 2005). Recognized as a master gene for chondrogenesis, SOX9 is expressed in all cartilage progenitors (Sahar et al., 2005; Thomas et al., 1997), and studies show that all osteochondrogenic cells derive from SOX9-expressing progenitors (Akiyama et al., 2005). In our TNCC cultures, FGF8 increased SOX9 expression 44-fold, making it the fourth most expressed transcript among hundreds analyzed. Importantly, FGF8 activates SOX9, as shown in Xenopus assays (Monsoro-Burq et al., 2003). Considering that SOX9 regulates COL2A1 expression (Cheng and Genever, 2010), the 2.47-fold increase in the cartilage-specific gene COL2A1 transcripts in our FGF8-treated cultures may result from this upregulation in SOX9 expression.
During migration, SOX9 and the transcription factor MSX2 are co-expressed in a subpopulation of CNCCs to form the mandible (Nelms and Labosky, 2010). SOX9 indicates chondrogenic lineage determination, while MSX2 inhibits chondrogenic differentiation until CNCCs complete their migration (Takahashi et al., 2001). We found that MSX2 levels increase by 2.98-fold in TNCCs treated with FGF8, suggesting a pro-chondrogenic differentiation program is kept on “stand-by” due to high MSX2 levels, which helps explain how SOX9 promotes multipotency instead of chondrogenesis.
SOX10, another member of the high-mobility group (HMG) gene family, functions by inhibiting neuronal differentiation while preserving the potential for neural lineage in rats. Therefore, it plays a key role in maintaining stem or progenitor cells and contributes to peripheral gliogenesis (Kim et al., 2003). In chicken embryos, overexpression of SOX10 increases the migration of cells from the neural tube, but these cells remain undifferentiated (McKeown et al., 2005). Notably, SOX10 and SOX9 mutually regulate each other and activate COL2A1 expression during cartilage differentiation from NCCs (Suzuki et al., 2006). In our cultures, FGF8 treatment increased SOX10 expression by 3.6-fold, suggesting that this transcription factor may also help maintain TNCCs in an undifferentiated and multipotent state.
Moreover, upstream to SOX10 and SOX9, we found another essential element in this circuitry: the transcription factor PAX3. PAX3 interacts with TGFB2 in a coordinated gene regulatory network to keep NCCs undifferentiated, mediated by downstream effector genes, including SOX9 (Nakazaki et al., 2009). Interestingly, in our FGF8-treated cultures, PAX3 and TGFB2 increased 3.25-fold and 2.0-fold, respectively. In Xenopus laevis NC induction, FGF8 activates MSX1, which activates PAX3 upstream to SOX9, SOX10, and another critical transcription factor discussed below: FOXD3 (Monsoro-Burq et al., 2003; Nelms and Labosky, 2010; Sato et al., 2005).
FOXD3 is a winged-helix transcription factor and an early marker of the NC lineage (Nelms and Labosky, 2010). Mutant embryos lacking FOXD3 struggle to maintain the NC progenitor pool during pre-migratory and early migratory stages, leading to defects in craniofacial skeleton development and the near-complete absence of the peripheral and enteric nervous systems (Teng et al., 2008). FOXD3 is crucial for preserving the multipotency and self-renewal of embryonic stem (ES) cells and trophoblast stem cells in culture (Hanna et al., 2002; Liu and Labosky, 2008; Tompers et al., 2005). In Zebrafish, single-cell studies show that FOXD3 exerts bimodal activity in NCCs, acting as a switch from "permissive" to "repressive" nucleosome and chromatin organization to maintain multipotency and define cell fates, similar to SOX9 and SOX10 (Lukoseviciute et al., 2018). Notably, data from Mundell and Labosky indicated that FOXD3 is necessary to prevent myofibroblast differentiation and to maintain NCSCs in a multipotent state (Mundell and Labosky, 2011). Additionally, SOX10 expression is significantly reduced in FOXD3-null mutant cells, underscoring their interdependence. Thus, FOXD3 acts as a gatekeeper for multipotent NCSCs, repressing mesenchymal lineages while preserving neural fates and multipotency (Mundell and Labosky, 2011).
Some studies also suggest that FOXD3 and SOX9 act together (Cheung and Briscoe, 2003). For example, their expression largely overlaps during NC specification in frogs (Martik and Bronner, 2017). Recent multiplex spatial genomics analysis in chicken embryos revealed an unbiased hierarchical clustering of SOX9 and FOXD3 genes in pre-migratory NCCs, suggesting they act as co-binding factors in the gene regulatory network (Lignell et al., 2017). This relationship can help to explain the early requirement for FOXD3 in the repression of ectomesenchymal fates shortly after cells leave the neural tube, before arrival at the pharyngeal arches or cardiac outflow tract. Loss of FOXD3 in the NC resulted in ectopic activation of both SOX9 and RUNX2 in cranial NC; therefore, premature endochondral bone and cartilage formation may underlie the severe craniofacial defects observed in FOXD3 NC mutant embryos (Mundell and Labosky, 2011).
Importantly, in Xenopus, FGF8 mRNA injections induce the expression of FOXD3 in animal cap assays (Monsoro-Burq et al., 2003). In our cultures, FOXD3 transcripts were expressed 4.4-fold higher in FGF8-treated cultures, evidencing that the growth factor somehow regulates FOXD3 expression.
Taking all this data together, we propose that FGF8 can maintain the progenitor status of TNCCs by upregulating several transcription factors (MSX1, MSX2, SOX9, SOX10, PAX3, and FOXD3) related to undifferentiation and multipotency. FOXD3 may function at a crucial early step in NCSC multipotency by maintaining responsiveness to FGF8 signaling and functions as a gatekeeper avoiding SOX9 pro-chondrogenic activation. In this sense, FOXD3 acts similarly (and perhaps in conjunction) to MSX2 by repressing the chondrogenic differentiation program elicited by SOX9. In a second moment, SOX9 must keep its higher expression, but the downregulation of FOXD3 and MSX2 is required for the mesenchymal program to occur. This effect elicited by FGF8 can follow a classical hierarchical model or, eventually, a hub model as proposed by Azambuja and Simoes-Costa (2021); (Fig. 3).
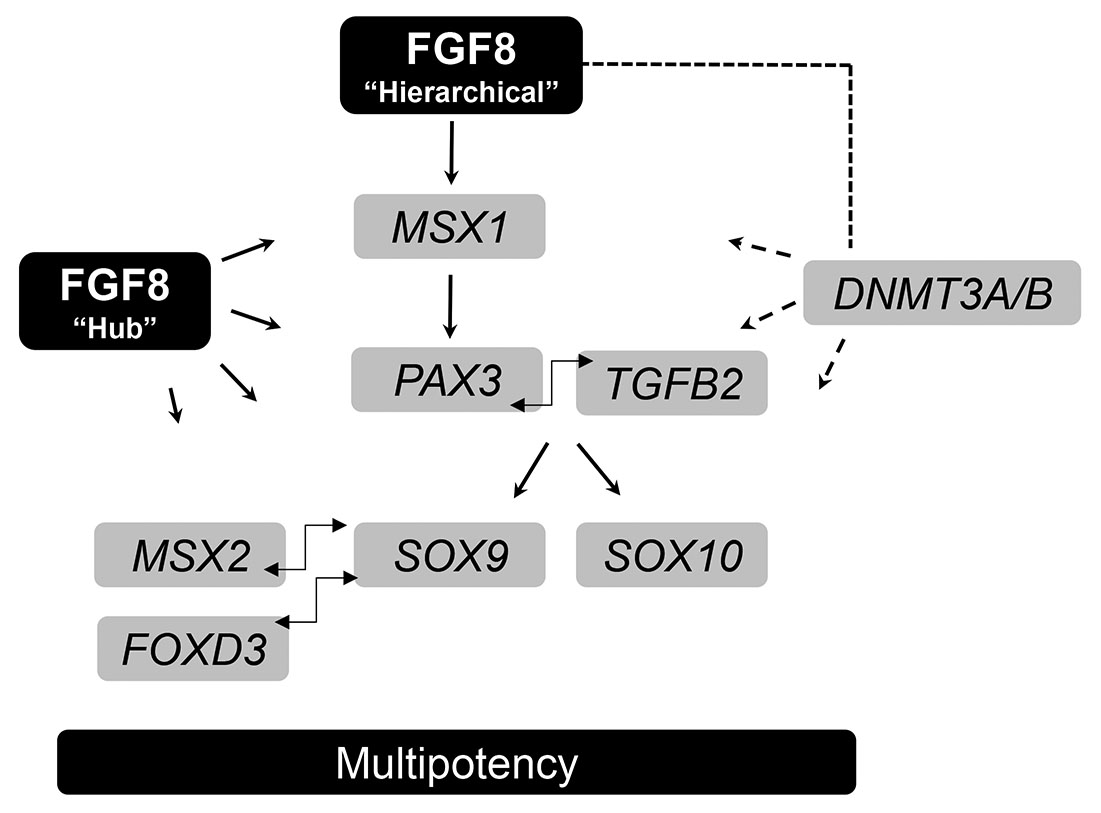
Fig. 3. A possible gene regulatory network (GRN) that can sustain trunk neural crest (TNCC) multipotency.
In a hierarchical model, FGF8 initially promotes the increase of MSX1, which sequentially activates PAX3. PAX3, in turn, activates SOX9 and SOX10, and SOX9 subsequently activates FOXD3. The concomitant high expression of MSX2, FOXD3, and SOX9 may be necessary to keep TNCCs in a multipotent state instead of differentiating into chondrocytes. The downregulation of MSX2 and FOXD3 is required for TNCCs to undergo chondrogenesis. In a hub model, FGF8 acts directly on some (or possibly all) of the elements of the GRN. Several studies in different animal models have shown that FGF8 promotes the upregulation of most of these genes. A possible mechanism of FGF8 action is indirect, through activation of DNA methyltransferases (DNMT3A and DNMT3B), as both were highly upregulated in FGF8-treated TNCC cultures.
The upregulation of certain transcription factors associated with undifferentiation and stem cell maintenance does not confirm that FGF8 promotes multipotentiality, as these transcription factors are also involved in lineage determination, which opposes undifferentiation and multipotentiality. Importantly, undifferentiation is not synonymous with multipotency. Multipotency is a latent property that can be revealed only by exposing stem or progenitor cells to conditions that elicit overt differentiation. Therefore, the question is whether we have some evidence relating FGFs (including FGF8) to sustain NC in a multipotent status.
Our research group has long explored the multipotentiality of NCCs through clonal analysis. Most TNCCs during FGF2 treatment remained unmarked for all expected lineage makers, indicating that FGF2 may inhibit their differentiation (Bittencourt et al., 2013). Additionally, FGF2 increased the proportion of tetra- and tri-potent progenitors in clonal cultures by at least two-fold and promoted self-renewal of glial-fibroblast (GF) progenitors (Bittencourt et al., 2013). Moreover, in clonal cultures treated with FGF8 during primary cultures (15 h) and with FGF8 + SHH in secondary cultures (during 48 h) the number of highly multipotent (hexa, penta, and tetrapotent) corresponded to 53% of total progenitors, compared to only 13.5% in FGF8-untreated cultures (da Costa et al., 2018). These data point to both FGF2 and FGF8 promoting the multipotentiality of NCCs.
Our data align with Shao et al., (2015), which show that CNC-derived mesenchymal cells from Wnt1cre; Rosa26R-FGF8 mice embryos can maintain their progenitor status and multipotency. The study found that these cells proliferated until passage 16 and retained their differentiation capabilities up to passage 10. When differentiation was induced, FGF8-overexpressing cells at passage 10 showed the same abilities as those at passage 3 in terms of undergoing adipogenesis, chondrogenesis, and neurogenesis. Extremely important, at passage 7, wild-type cells treated with exogenous FGF8 (50 ng/mL) exhibited similar or enhanced differentiation capabilities compared to untreated cells from passage 3. This suggests that exogenous FGF8 supports the progenitor status and multipotency of CNC-derived mesenchymal cells in vitro.
We propose that FGF8 influences differential chromatin states through epigenetic mechanisms, potentially via de novo DNA methyltransferases (DNMTs) (Cheng and Blumenthal, 2008). DNMT3A, in particular, is highly expressed at the neural plate border during gastrulation and later in migrating NCCs (Hu et al., 2012). It serves as a molecular switch, deactivating neural tube transcription factors and enabling the expression of genes that promote NC cell identity. The knocking down of DNMT3A reduces the levels of critical genes like FOXD3, SOX9, SOX10, and ETS1 in chick embryos (Hu et al., 2012). In our quail FGF8-treated TNCC cultures, DNMT3A expression increased by 4.47-fold, suggesting that FGF8 may regulate key NC transcription factors through upregulation of DNMT3A.
FGF8 treatment also resulted in an 11.23-fold increase in DNMT3B transcripts. Interestingly, knocking down DNMT3B upregulates essential genes like TFAP2A, SOX9, SOX10, SNAIL2, and FOXD3, which contradicts our findings (Hu et al., 2014). Since epigenetic regulation is complex, more studies are needed to understand the interactions between FGF8 and DNMTs. If epigenetic mechanisms influence FGF8's effects on TNCC chondrogenesis, DNMTs are likely candidates based on our transcriptome results.
In sum, in the first scenario, we propose that FGF8, eventually acting through an epigenetic mechanism, can keep NCCs multipotent in an undifferentiated state until they find a suitable environment to express their full potential, which includes the chondrogenic one.
FGF8 gives TNCCs a cranial identity
In 2002, Imelda McGonnell and Anthony Graham were the first to report that avian TNCCs could differentiate into bone and cartilage when placed in a suitable environment (McGonnell and Graham, 2002). However, no mechanism was proposed by the authors to explain their observations. The following year, Abzhanov and colleagues observed skeletogenic differentiation in TNCCs from the sacral region of chick embryos. After 14 days of culture, real-time PCR analysis showed high expression of cranial genes ID2 and NOELIN1, while the trunk marker HOXB4 was down-regulated. The researchers suggested that chondrogenesis occurs after a prolonged culture period, allowing TNCCs to down-regulate HOXB4 and adopt characteristics of CNCCs (Abzhanov et al., 2003).
The study of Ido and Ito, 2006, is relevant to our work as they treated mice TNCCs with FGF2. After three days, they observed a significant downregulation of HOX9 expression. This indicates that FGF2 may promote chondrogenesis by reducing HOX9 levels. However, by days five and ten, the percentage of HOX9-expressing cells in FGF2-treated cultures was similar to that in untreated cultures. Thus, unlike Abzhanov’s findings, long-term cultures are unnecessary for chondrocyte development, and permanent downregulation of HOX genes is not essential for chondrogenesis (Ido and Ito, 2006).
We detected HOXD9 exclusively in TNC cultures treated with FGF8, suggesting that HOXD9 may be related to FGF8-promoting chondrogenesis. HOXD9 is crucial for limb chondrogenesis; exposure of rat embryo hindlimb bud mesenchymal cells (rEHBMCs) to all-trans-retinoic acid (ATRA) for 24 hours suppresses chondrogenesis by inhibiting HOXD9 and its targets like SOX9 and COL2A1 (Hong et al., 2021). Therefore, a possible mechanism is that FGF8 enhances HOXD9 expression, leading to a significant increase in SOX9 (44-fold) and upregulation of COL2A1 (2.47-fold) in treated cultures.
An interesting aspect of this puzzle is the expression of the cranial-specific genes in the trunk neural crest (Fig. 2C). For example, ID2 is a cranial-specific gene detected in both Abzhanov’s long-term TNC cultures and by Ito and Ido in mouse FGF2-treated TNC cultures (Abzhanov et al., 2003; Ido and Ito, 2006). ID2 was upregulated by FGF2 treatment in TNCCs on day 5, but by day 10, its levels were similar, regardless of whether FGF2 was present. We also detected ID2 in our TNCC transcriptome, although FGF8 treatment did not affect its levels.
Another cranial-specific gene detected exclusively in FGF8-treated TNC cultures was the LIM Homeobox Protein 8 (LHX8), whose expression is restricted to face mesenchyme, including pharyngeal arches and clefts, and plays a crucial role in tooth formation (Zhou et al., 2015). This suggests a possible shift from trunk to cranial NCC identity, which may stem from the culture conditions rather than FGF signaling, as our FGF8-untreated cultures also showed chondrogenesis.
Finally, of fundamental importance is the study of Simoes-Costa and Bronner, which identified a cranial-specific gene regulatory network crucial for ectomesenchyme development (Simoes-Costa and Bronner, 2016). They manipulated NC identity by electroporating expression constructs into the trunk neural tube of HH10 embryos. Transfection with cranial-specific factors—SOX8, TFAP2b, and ETS1—activated skeletogenic genes like RUNX2 and ALX1 in TNCCs. These reprogrammed TNCCs developed chondrogenic potential and formed ectopic cartilage nodules in chicken embryos. Notably, in our TNCC cultures treated with FGF8, TFAP2b expression increased 3.42-fold, and ETS1 increased 2.18-fold. Despite augmented in FGF8-treated cultures, the expression of the SOX8 gene was not statistically significant compared to untreated cultures (Fig. 2C).
More recently, a transcriptional analysis of reprogramed TNCCs showed that more genes are shared between reprogramed TNCCs and cranial NC than with TNCCs. Key enriched genes included SOX9, COL2A1, MSX1, and MSX2, which were also elevated in our FGF8-treated cultures (Marable and Bronner, 2023). However, the transcriptional profile of reprogrammed TNCCs is not identical to the cranial NC (Marable and Bronner, 2023). Therefore, is essential to mention that “cranial-like” indicates the ability of the reprogrammed TNCCs to form cartilage.
What seems most surprising is the fact that the overexpression of a few key transcription factors can modulate NC axial specification and differentiation. Different environmental conditions can elicit the overexpression of these genes. In line with this, we must consider that even FGF8-untreated TNC cultures display chondrogenesis. The fact that ID2, TFAP2b, and ETS1 are expressed in both untreated and FGF8-treated cultures indicates that the mere removal of TNCCs from the in vivo environment is sufficient (and necessary) to promote a shift from trunk to "cranial-like identity". We propose that FGF8 acts amplifying this phenomenon by upregulating some key genes like TFAP2b, ETS1, SOX9, COL2A1, and also COL1A2 (a bone marker), reinforcing their crucial role in giving rise to ectomesenchyme as demonstrated by Simoes and Bronner in 2016. It is tempting to speculate if a differential spatial/temporal expression of FGF8 in some target tissues during development could explain the contribution of TNCCs to mesenchymal derivatives in some animal species.
Conclusion
We propose that mesenchymal potential is an ancient TNCC feature that can be unveiled under particular environmental conditions. It is rarely and sparsely detected in vivo in some fish, turtles and, more recently, armadillos. Independently of the scenario proposed, the present work's results highlight that removing TNCCs from some in vivo environmental restrictions allows their chondrogenic potential to be expressed. We propose that FGF8, possibly acting through an epigenetic mechanism (upregulating DNMT3A and B), may help keep NCCs multipotent in an undifferentiated state until they find a suitable environment to express their full potential, which includes the chondrogenic potential. In the second scenario, which does not necessarily exclude the first, TNC cultures allow a trunk shift to a “cranial-like” identity by expressing some cranial-specific genes, like ID2, TFAP2b, and ETS1. FGF8 potentiates this phenomenon by upregulating some (i.e., TFAP2b and ETS1) and others associated with chondrogenesis (SOX9, COL2A1).
Materials and Methods
Ethical concerns
The animal protocol used in this work was approved by the Ethics Committee on Animal Use of the Federal University of Santa Catarina (CEUA/UFSC) under the protocol n˚ 7224130916 (ID 000289).
TNCC cultures
TNCCs were isolated from explanted neural tubes at the thoracic level (last ten somites) from quail embryos at the 18-24 somite stage. Briefly, the explanted neural tubes were seeded on a 35 mm plastic dish (Corning®) with maintenance medium consisting of alpha-modified minimum essential medium (α-MEM, Invitrogen), enriched with 10% fetal bovine serum (FBS, Invitrogen®) and 2% chicken embryo extract. Additionally, the medium was supplemented or not with 100 ng/ml of FGF8 (recombinant Human FGF8-8b, R&D Systems). After 15 hours of primary culture and TNCC emigration from the explanted tissue, the neural tubes were discarded under microscopic control with tungsten needles, and the migrated TNCCs were harvested with trypsin 0.05% (Invitrogen®) to perform secondary cultures or transcriptomic analysis (see below).
Isolated TNCCs were seeded in 96-well plates covered with 20 µl Matrigel (Corning®) at a density of 400 cells/well and maintained in medium supplemented or not with 10 or 100 ng/ml of FGF8 for the duration of the secondary cultures, i.e., ten days, with the medium replaced twice a week.
Chondrocyte detection
After ten days of secondary cultures, TNCCs were fixed with 4% paraformaldehyde for 40 min. Chondrocytes were first evidenced as three-dimensional cell aggregates under phase-contrast microscopy and detected by immunofluorescence to Chondroitin Sulfate (CS) (1:1600; clone CS56 Sigma®) with a specific secondary antibody, Alexa-488 (Invitrogen®). Cell nuclei were stained with 4’,6-Diamino-2-Phenylindole (DAPI). Fluorescence was observed with an Olympus IX71 microscope.
Phenotypic Statistical analysis of TNC cultures
Phenotypic analysis was used to compare the mean number of cartilage nodules using Student’s two-tailed t-test. The frequencies of cartilage nodules were analyzed using the Chi-square test (X2). Statistical analyses were performed with Prism GraphPad Software®.
Library construction and RNA sequencing
The library construction and next-generation sequencing methods have been previously published (Marcon et al., 2020), and the data generated were utilized for the expression analyses presented here. In summary, 1 μg of total RNA was used for cDNA library preparation from three independent replicates of biological samples (i.e., 15 hours migrating TNCCs treated or not with 100 ng/ml of FGF8). Following the manufacturer's instructions, the library was prepared using the TruSeq Stranded Total RNA Sample Preparation kit (Illumina®, Inc., San Diego, CA, USA). The purified products were evaluated with an Agilent Bioanalyzer (Agilent®, Santa Clara, CA, USA). According to the manufacturer's recommendations, the high-throughput sequencing was performed in an Illumina HiSeq 2500 platform using the TruSeq SBS Kit v3—HS (Illumina®, Inc., San Diego, CA, USA).
Bioinformatic analysis
Data analysis for mapping and counting was performed using the R package Rsubread with the new version of the Coturnix japonica 2.1 genome (Bridges et al., 2009). Parameters were set for the unique mapping of reads. Non-expressed and weakly expressed genes were defined as having ≤1 count (read) per million and were excluded from the differential expression analysis. Differential gene expression analysis was conducted to compare normalized and filtered read counts between FGF8 untreated cells (-FGF8) and FGF8 treated cells (+FGF8), using the Bioconductor package edgeR (v. 3.20.9). Setting a p-value of ≤0.05 as statistically significant, and considering a log2 fold change (|log2FC|) ≥1. All Bioinformatic statistical and graph representations were performed using R (v. 3.5.2).
Data availability
The RNA-seq raw data are deposited in the NCBI GEO repository under the accession number PRJNA1148439.
Supplementary Material
Acknowledgements
The authors would like to thank LAMEB-UFSC, Carlos Chagas Institute, and the Program for Technological Development in Tools for Health-RTP-FIOCRUZ for technical support and for providing their respective infrastructure for carrying out some of the experimental tests. This work was supported by the Ministério da Ciência e Tecnologia/Conselho Nacional de Desenvolvimento Científico e Tecnológico (MCT/ CNPq/Brazil), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil), and Fundação de Amparo de Pesquisa do Estado de Santa Catarina (FAPESC, SC, Brazil).
Declarations
Conflicts of interest
The author(s) declared no potential conflicts of interest concerning the research, authorship, commercial relationships, and publication.
Competing interests
The authors declare that no competing interests exist.
References
Abzhanov A., Cordero D. R., Sen J., Tabin C. J., Helms J. A. (2007). Cross–regulatory interactions between Fgf8 and Shh in the avian frontonasal prominence. Congenital Anomalies 47: 136-148.
Abzhanov A., Tabin C. J. (2004). Shh and Fgf8 act synergistically to drive cartilage outgrowth during cranial development. Developmental Biology 273: 134-148.
Abzhanov A., Tzahor E., Lassar A. B., Tabin C. J. (2003). Dissimilar regulation of cell differentiation in mesencephalic (cranial)and sacral (trunk) neural crest cells in vitro. Development 130: 4567-4579.
Akiyama H., Kim J.E., Nakashima K., Balmes G., Iwai N., Deng J. M., Zhang Z., Martin J. F., Behringer R. R., Nakamura T., de Crombrugghe B. (2005). Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proceedings of the National Academy of Sciences 102: 14665-14670.
Azambuja A. P., Simoes-Costa M. (2021). The connectome of neural crest enhancers reveals regulatory features of signaling systems. Developmental Cell 56: 1268-1282.e6.
Bittencourt D. A., da Costa M. C., Calloni G. W., Alvarez-Silva M., Trentin A. G. (2013). Fibroblast Growth Factor 2 Promotes the Self-Renewal of Bipotent Glial Smooth Muscle Neural Crest Progenitors. Stem Cells and Development 22: 1241-1251.
Bridges S. M., Burgess S. C., McCarthy F. M. (2009). Introduction to the Proceedings of the Avian Genomics and Gene Ontology Annotation Workshop. BMC Genomics 10: I1.
Calloni G. W., Glavieux-Pardanaud C., Le Douarin N. M., Dupin E. (2007). Sonic Hedgehog promotes the development of multipotent neural crest progenitors endowed with both mesenchymal and neural potentials. Proceedings of the National Academy of Sciences 104: 19879-19884.
Calloni G. W., Le Douarin N. M., Dupin E. (2009). High frequency of cephalic neural crest cells shows coexistence of neurogenic, melanogenic, and osteogenic differentiation capacities. Proceedings of the National Academy of Sciences 106: 8947-8952.
Cheng A., Genever P. G. (2010). SOX9 determines RUNX2 transactivity by directing intracellular degradation. Journal of Bone and Mineral Research 25: 2680-2689.
Cheng X., Blumenthal R. M. (2008). Mammalian DNA Methyltransferases: A Structural Perspective. Structure 16: 341-350.
Cheung M., Briscoe J. (2003). Neural crest development is regulated by the transcription factor Sox9. Development 130: 5681-5693.
Creuzet S., Schuler B., Couly G., Le Douarin N. M. (2004). Reciprocal relationships between Fgf8 and neural crest cells in facial and forebrain development. Proceedings of the National Academy of Sciences 101: 4843-4847.
da Costa M. C., Trentin A. G., Calloni G. W. (2018). FGF8 and Shh promote the survival and maintenance of multipotent neural crest progenitors. Mechanisms of Development 154: 251-258.
Degistirici O., Jaquiery C., Schönebeck B., Siemonsmeier J., Götz W., Martin I., Thie M. (2008). Defining Properties of Neural Crest–Derived Progenitor Cells from the Apex of Human Developing Tooth. Tissue Engineering Part A 14: 317-330.
Gilbert S. F., Bender G., Betters E., Yin M., Cebra-Thomas J. A., (2007). The contribution of neural crest cells to the nuchal bone and plastron of the turtle shell. Integrative and comparative biology 47: 401-408.
Hackland J. O.S., Shelar P. B., Sandhu N., Prasad M. S., Charney R. M., Gomez G. A., Frith T. J.R., García-Castro M. I. (2019). FGF Modulates the Axial Identity of Trunk hPSC-Derived Neural Crest but Not the Cranial-Trunk Decision. Stem Cell Reports 12: 920-933.
Hanna L. A., Foreman R. K., Tarasenko I. A., Kessler D. S., Labosky P. A. (2002). Requirement for Foxd3 in maintaining pluripotent cells of the early mouse embryo. Genes & Development 16: 2650-2661.
Hong Q., Li X.D., Xie P., Du S.X. (2021). All-trans-retinoic acid suppresses rat embryo hindlimb bud mesenchymal chondrogenesis by modulating HoxD9 expression. Bioengineered 12: 3900-3911.
Hu N., Strobl-Mazzulla P. H., Simoes-Costa M., Sánchez-Vásquez E., Bronner M. E. (2014). DNA methyltransferase 3B regulates duration of neural crest production via repression of Sox10. Proceedings of the National Academy of Sciences 111: 17911-17916.
Hu N., Strobl-Mazzulla P., Sauka-Spengler T., Bronner M. E. (2012). DNA methyltransferase3A as a molecular switch mediating the neural tube-to-neural crest fate transition. Genes & Development 26: 2380-2385.
Ido A., Ito K. (2006). Expression of chondrogenic potential of mouse trunk neural crest cells by FGF2 treatment. Developmental Dynamics 235: 361-367.
Kague E., Gallagher M., Burke S., Parsons M., Franz-Odendaal T., Fisher S. (2012). Skeletogenic Fate of Zebrafish Cranial and Trunk Neural Crest. PLoS ONE 7: e47394.
Kim J., Lo L., Dormand E., Anderson D. J. (2003). SOX10 Maintains Multipotency and Inhibits Neuronal Differentiation of Neural Crest Stem Cells. Neuron 38: 17-31.
Krmpotic C. M., Nishida F., Galliari F. C., Pombo M. T., Acuña F., Barbeito C. G., Carlini A. A. (2021). The Dorsal Integument of the Southern Long-Nosed Armadillo Dasypus hybridus (Cingulata, Xenarthra), and a Possible Neural Crest Origin of the Osteoderms. Discussing Evolutive Consequences for Amniota. Journal of Mammalian Evolution 28: 635-645.
Le Douarin N., Kalcheim C., (1999). The Neural Crest. Cambridge University Press, Cambridge.
Lignell A., Kerosuo L., Streichan S. J., Cai L., Bronner M. E. (2017). Identification of a neural crest stem cell niche by Spatial Genomic Analysis. Nature Communications 8: 1830.
Liu Y., Labosky P. A. (2008). Regulation of Embryonic Stem Cell Self-Renewal and Pluripotency by Foxd3. Stem Cells 26: 2475-2484.
Lukoseviciute M., Gavriouchkina D., Williams R. M., Hochgreb-Hagele T., Senanayake U., Chong-Morrison V., Thongjuea S., Repapi E., Mead A., Sauka-Spengler T. (2018). From Pioneer to Repressor: Bimodal foxd3 Activity Dynamically Remodels Neural Crest Regulatory Landscape In Vivo. Developmental Cell 47: 608-628.e6.
Marable S. S., Bronner M. E. (2023). Reprogramming of trunk neural crest to a cranial crest-like identity alters their transcriptome and developmental potential. Differentiation 131: 27-37.
Marcon B. H., Spangenberg L., Bonilauri B., Robert A. W., Angulski A. B. B., Cabo G. C., Cofré A. R., Bettes P. S. L., Dallagiovanna B., Shigunov P. (2020). Data describing the experimental design and quality control of RNA-Seq of human adipose-derived stem cells undergoing early adipogenesis and osteogenesis. Data in Brief 28: 105053.
Martik M. L., Bronner M. E. (2017). Regulatory Logic Underlying Diversification of the Neural Crest. Trends in Genetics 33: 715-727.
McGonnell I. M., Graham A. (2002). Trunk Neural Crest Has Skeletogenic Potential. Current Biology 12: 767-771.
McKeown S. J., Lee V. M., Bronner‐Fraser M., Newgreen D. F., Farlie P. G. (2005). Sox10 overexpression induces neural crest‐like cells from all dorsoventral levels of the neural tube but inhibits differentiation. Developmental Dynamics 233: 430-444.
Monsoro-Burq A.H., Fletcher R. B., Harland R. M. (2003). Neural crest induction by paraxial mesoderm in Xenopus embryos requires FGF signals. Development 130: 3111-3124.
Monsoro-Burq A.H., Wang E., Harland R. (2005). Msx1 and Pax3 Cooperate to Mediate FGF8 and WNT Signals during Xenopus Neural Crest Induction. Developmental Cell 8: 167-178.
Mundell N. A., Labosky P. A. (2011). Neural crest stem cell multipotency requires Foxd3 to maintain neural potential and repress mesenchymal fates. Development 138: 641-652.
Nakamura H., Lievre C. S. A.L. (1982). Mesectodermal capabilities of the trunk neural crest of birds. Development 70: 1-18.
Nakazaki H., Shen Y.W., Yun B., Reddy A., Khanna A., Mania-Farnell B., Ichi S., Mclone D. G., Tomita T., Mayanil C. S. K. (2009). Transcriptional regulation by Pax3 and TGFbeta2 signaling: a potential gene regulatory network in neural crest development. The International Journal of Developmental Biology 53: 69-79.
Nelms B. L., Labosky P. A. (2010). Transcriptional Control of Neural Crest Development. Colloquium Series on Developmental Biology 1: 1-227.
Noden D. M. (1978). The control of avian cephalic neural crest cytodifferentiation. Developmental Biology 67: 296-312.
Prasad M. S., Charney R. M., García‐Castro M. I. (2019). Specification and formation of the neural crest: Perspectives on lineage segregation. Genesis 57: e23276.
Ramos-Hryb A. B., Da-Costa M. C., Trentin A. G., Calloni G. W. (2013). Matrigel supports neural, melanocytic and chondrogenic differentiation of trunk neural crest cells. The International Journal of Developmental Biology 57: 885-890.
Rodrigues-Da-Silva M. A., de Espindola da Silveira G., Taufer C. R., Calloni G. W. (2022). The mesenchymal potential of trunk neural crest cells. The International Journal of Developmental Biology 66: 317-331.
Sahar D. E., Longaker M. T., Quarto N. (2005). Sox9 neural crest determinant gene controls patterning and closure of the posterior frontal cranial suture. Developmental Biology 280: 344-361.
Sato T., Sasai N., Sasai Y. (2005). Neural crest determination by co-activation of Pax3 and Zic1 genes in Xenopus ectoderm. Development 132: 2355-2363.
Shao M., Liu C., Song Y., Ye W., He W., Yuan G., Gu S., Lin C., Ma L., Zhang Y., Tian W., Hu T., Chen Y.P. (2015). FGF8 signaling sustains progenitor status and multipotency of cranial neural crest-derived mesenchymal cells in vivo and in vitro. Journal of Molecular Cell Biology 7: 441-454.
Simoes-Costa M., Bronner M. E. (2016). Reprogramming of avian neural crest axial identity and cell fate. Science 352: 1570-1573.
Suzuki T., Sakai D., Osumi N., Wada H., Wakamatsu Y. (2006). Sox genes regulate type 2 collagen expression in avian neural crest cells. Development, Growth & Differentiation 48: 477-486.
Takahashi K., Nuckolls G.H., Takahashi I., Nonaka K., Nagata M., Ikura T., Slavkin H.C., Shum L. (2001). Msx2 is a repressor of chondrogenic differentiation in migratory cranial neural crest cells†. Developmental Dynamics 222: 252-262.
Taufer C. R., Rodrigues-Da-Silva M. A., Calloni G. W. (2020). PuraMatrix allows differentiation of a broad repertoire of neural and mesenchymal phenotypes from trunk neural crest. The International Journal of Developmental Biology 64: 433-443.
Techawattanawisal W., Nakahama K., Komaki M., Abe M., Takagi Y., Morita I. (2007). Isolation of multipotent stem cells from adult rat periodontal ligament by neurosphere-forming culture system. Biochemical and Biophysical Research Communications 357: 917-923.
Teng L., Mundell N. A., Frist A. Y., Wang Q., Labosky P. A., (2008). Requirement for Foxd3 in the maintenance of neural crest progenitors. Development (Cambridge, England) 135: 1615-1624.
Thomas B. L., Tucker A. S., Qiu M., Ferguson C. A., Hardcastle Z., Rubenstein J. L. R., Sharpe P. T. (1997). Role of Dlx-1 and Dlx-2 genes in patterning of the murine dentition. Development 124: 4811-4818.
Tompers D. M., Foreman R. K., Wang Q., Kumanova M., Labosky P. A. (2005). Foxd3 is required in the trophoblast progenitor cell lineage of the mouse embryo. Developmental Biology 285: 126-137.
Trainor P. A., Ariza-McNaughton L., Krumlauf R. (2002). Role of the Isthmus and FGFs in Resolving the Paradox of Neural Crest Plasticity and Prepatterning. Science 295: 1288-1291.
Zhao M., Isom S. C., Lin H., Hao Y., Zhang Y., Zhao J., Whyte J. J., Dobbs K. B., Prather R. S. (2009). Tracing the Stemness of Porcine Skin-Derived Progenitors (pSKP) Back to Specific Marker Gene Expression. Cloning and Stem Cells 11: 111-122.
Zhou C., Yang G., Chen M., Wang C., He L., Xiang L., Chen D., Ling J., Mao J. J. (2015). Lhx8 mediated Wnt and TGFβ pathways in tooth development and regeneration. Biomaterials 63: 35-46.