Int. J. Dev. Biol. 67: 27 - 37 (2023)
Importance of the filamin A-Sav1 interaction in organ size control: evidence from transgenic mice
Original Article | Published: 1 August 2023
Abstract
The nucleocytoplasmic translocation of yes-associated protein 1 (YAP1) controls the growth of animal tissues and organs. YAP1 binds to transcription factors in the nucleus to activate the transcription of proliferation and anti-apoptotic genes. The Hippo pathway prevents the nuclear translocation of YAP1 by phosphorylating YAP1, while mechanical forces promote it by opening the nuclear pore complex and stimulating other signaling pathways. Recently we found that Protein salvador homolog 1 (SAV1), a component of the Hippo pathway, interacts with filamin A (FLNA) in a force-dependent manner, raising a possibility that the Hippo pathway is regulated by mechanical force through the FLNA-SAV1 axis. To test this hypothesis, we generated conditional knock-in (KI) mice expressing non-Flna-binding mutant Sav1 in hepatocytes by crossing with mice carrying Cre recombinase driven by the serum albumin (alb) gene promoter. Unexpectedly, the insertion of the flox cassette skipped exon 2, resulting in a shorter Sav1 in all the transgenic mice. Since exon 2 encodes a fragment containing a Flna-binding domain, we analyzed both point mutant KI and exon 2-deleted mutant mice. Here we show that disruption of the Flna-Sav1 interaction in the mouse liver promotes apoptosis and suppresses tissue and organ growth without affecting the phosphorylation level of Yap1. These results provide evidence that the growth of animal tissues and organs is regulated by apoptosis downstream of the force-dependent FLNA-SAV1 interaction, at least in part.
Keywords
Filamin A, SAV1, mechanotransduction, Hippo pathway, YAP1, hepatocyte, knock-in mouse, organ size
Introduction
Translocation of YAP1 (or Yki in Drosophila) between the cytosol and nucleus is a key determinant of cell growth and survival (Finch-Edmondson and Sudol, 2016; Maehama et al., 2021; Piccolo et al., 2014; Zeng and Hong, 2008; Zhao et al., 2008). When YAP1 is located in the nucleus, YAP1 binds to transcription factors such as TEAD1 to activate transcription of proliferation and anti-apoptotic genes (Cheng et al., 2022; Zeybek et al., 2021; Zhou and Zhao, 2018; Zinatizadeh et al., 2021). Subcellular localization of YAP is controlled by multiple pathways (Russell and Camargo, 2022). One is the Hippo pathway which negatively regulates YAP retention in the nucleus. A key control step of the Hippo pathway is the localization and kinase activity of MST1/2 (or Hippo in Drosophila), which phosphorylates the downstream kinase LATS1/2 (Bae et al., 2017; Yin et al., 2013). The tumor suppressor protein SAV1 mediates the dimerization of MST to activate it and recruits LATS to the MST to promote the phosphorylation of LATS (Callus et al., 2006; Lin et al., 2020; Tran et al., 2020).
Phosphorylated LATS1/2 is active and in turn phosphorylates YAP1, leading to the cytoplasmic retention of phospho-YAP1 (Oh and Irvine, 2008). When the pathway is off, unphosphorylated YAP1 enters the nucleus to activate transcription. The second mechanism is mechanotransduction in which mechanical forces open the nuclear pore complex (NPC) to promote nuclear entry of YAP1 in a Hippo-independent manner (Das et al., 2016; Dupont et al., 2011; Elosegui-Artola et al., 2017; Kassianidou et al., 2019; Piccolo et al., 2014). However, since nuclear entry of non-phosphorylatable S127A mutant YAP1 is still sensitive to substrate rigidity, stretching of NPC is essential even for Hippo-dependent nuclear entry of YAP1. Moreover, another group showed that physiological mechanical strain at the apical membrane can reduce Hippo kinase dimerization to downregulate Hippo signaling, thereby activating Yki in Drosophila epithelia (Fletcher et al., 2018). The other mechanism includes non-proteolytic polyubiquitination of YAP by the SCFSKP2 E3 ligase complex (SKP2), which promotes YAP1 nuclear localization and transcriptional activity in independent of Hippo pathway-mediated phosphorylation of YAP (Yao et al., 2018). Tyrosine phosphorylation of YAP1 by Src also promotes its nuclear translocation and activation (Taniguchi et al., 2015).
Protein salvador homolog 1 (SAV1) is the scaffold protein that promotes the activation of MST-LATS kinase cascade in the Hippo pathway (de Amorim et al., 2021; Lin et al., 2020). SAV1 dimerization is essential for the trans-activation of MST (Lin et al., 2020) and loss of Sav1 (human protein names are designated with all letters in uppercase, whereas mouse protein names are with only the first letter in uppercase and the remaining letters in lowercase. Gene names are italicized) in mice induces liver enlargement, renal tubulointerstitial fibrosis, and senescence (Lee et al., 2010) (Leung et al., 2017; Seo et al., 2016). Consistent with these phenotypes, deletion of Mst1/2 in mouse hepatocytes results in enlarged livers due to excessive proliferation (Lu et al., 2010).
Recently, we found that SAV1 interacts with the mechanosensing domain of FLNA and disruption of the interaction promotes diffusion of SAV1 to the nucleus and cytoplasmic localization of YAP1 (Zhang et al., 2023). These results made us wonder if the Hippo pathway may be regulated by mechanical forces through the FLNA-SAV1 interaction. To test this hypothesis, we generated conditional KI mice expressing non-Flna-binding mutant Sav1 in hepatocytes by crossing with mice carrying Cre recombinase driven by alb gene promoter because previous research demonstrated that liver-specific deletion of Sav1 in mice results in increased liver size (Lee et al., 2010; Lu et al., 2010). We found that disruption of the Flna-Sav1 interaction in mouse liver promotes apoptosis and suppresses the growth of tissue and organ with unknown mechanisms. These results provide evidence that the growth of animal tissues and organs is regulated by apoptosis downstream of the force-dependent FLNA-SAV1 interaction at least in part.
Results
Generation of conditional mutant Sav1 knock-in C57BL/6 mice
To investigate if the FLNA-SAV1 interaction regulates animal organ size, we aimed to generate conditional KI mice expressing F117A (TTT to GCT) and T119A (ACA to GCA) (FT/AA) mutant Sav1 (corresponding to F116A and T118A mutations of human SAV1 that disrupt the interaction with FLNA (Zhang et al., 2023)) in liver using the albumin-Cre (alb-Cre)-lox system (Figs. 1B; S1). Since our previous data suggested that disruption of FLNA-SAV1 interaction suppresses YAP1 activity (Zhang et al., 2023), we speculated that non-Flna-binding mutation of Sav1 in whole tissue might be lethal for mice. If such mutation spoils the complete function of Sav1 (e.g. aggregation or degradation), hyperproliferation is expected because liver-specific knock-out (KO) of Sav1 results in enlargement of the liver due to excessive proliferation (Lee et al., 2010; Lu et al., 2010). To avoid the possibility of a lethal phenotype and complete disruption of Sav1 function, we generated liver-specific KI mice.
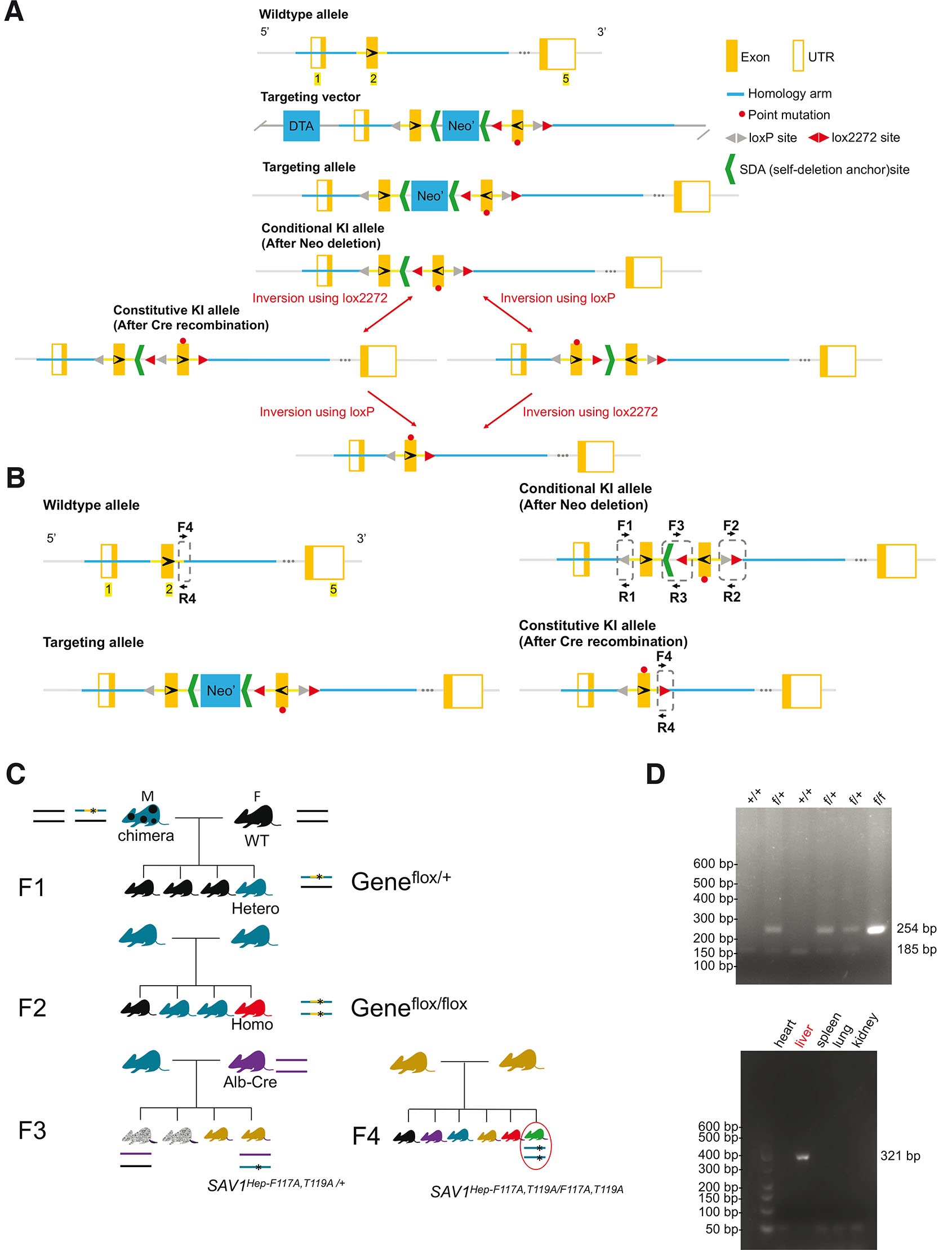
Fig. 1. Production of homologous recombinant embryonic stem (ES) cells to generate mutant SAV1 knock-in mice.
(A) Schematic representation of the Sav1 targeting strategy, showing the mouse Sav1 genomic locus, targeting vectors, and targeted locus. (B) Genotyping assay. Primers for targeted allele: loxP-F (F1) and loxP-R (R1). WT: 185 bp. Homozygotes: 254 bp. Heterozygotes: 254 bp/185 bp; lox2272-F (F2) and lox2272-R (R2). F3 and R3 are primers for confirmation of Neo deletion. Conditional KI allele: 260 bp; Primers for constitutive KI allele: cKI-F (F4) and cKI-R (R4). WT allele: 265 bp. Constitutive KI allele: 321 bp. (C) Breeding strategy for liver-specific mutant Sav1 Knock-in mouse. (D) Genomic DNA from mice tails was subjected to genotyping by PCR analysis. (D, top) The positions of products corresponding to the WT (+, 185 bp) and floxed (f, 254 bp) alleles were indicated. (D, bottom) Recombination PCR analysis of various organs of Sav1 Liv-cKI mice. The positions of products derived from the KI (321 bp) Sav1 alleles were indicated. Sav-1, salvador family WW domain containing protein 1.
Cre-negative heterozygous F1 mice (flox/+) were born healthy and fertile and developed normally. Then we tried to generate a Cre-negative homozygous F2 mouse (flox/flox). However, only two homozygotes (one male and one female) were found among 134 littermates, indicating that most of the homozygous F2 (flox/flox) mice were embryonic lethal. Although these two mice survived more than 10 months, they were small and infertile. Unfortunately, these mice died during the Covid-19 pandemic in 2020, and further analysis was not performed. Therefore, we altered the breeding strategy by crossing F1 with hepatocyte-specific alb-Cre mice and obtained liver-specific Cre-positive heterozygous F3 mouse (Sav1Hep-F116A, T118A/+) (Fig. 1C). Such Cre-positive heterozygous mice were born healthy and fertile and developed normally. To generate Cre-positive homozygous KI mice, Cre-positive heterozygous F3 mice were mated with each other. However, only two dwarf homozygotes were found among 90 littermates, indicating that most of the homozygous mice were embryonic lethal. Genotyping of WT mice and transgenic mice was confirmed and Cre recombination specifically occurred in the liver of Cre-positive mice as we expected (Fig. 1D).
Since the loss of Sav1 protein led to embryonic lethality in most mice (Lee et al., 2008) and since the superficial appearance of the homozygous flox/flox mice and the homozygous Cre positive Sav1Hep-F116A, T118A/Hep-F116A, T118A mice were observed to be similar, we decided to perform RT-PCR to evaluate the presence of Sav1 mRNA expressed in each mouse and western blotting to quantify the expression of Sav1 protein (Fig. 2). The RT-PCR results demonstrated that FT/AA mutation is successfully introduced in the Cre-positive flox/+ and flox/flox mice (Fig. 2C). Because the liver is a unique immunological organ with rich intrahepatic lymphocytes and diverse other cells (Racanelli and Rehermann, 2006), which do not express albumin to promote the expression of Cre recombinase, RT-PCR showed both KI and non-KI PCR products (Fig. 2B). Unexpectedly, the insertion of the flox cassette skips exon 2, resulting in shorter Sav1 in all transgenic mice, including (flox/+) (flox/+, Cre) and (flox/flox, Cre) mice (Fig. 2 B,C), which is missing FLNA-binding site. The point and exon 2-deleted Sav1 proteins were expressed in mouse tissue and tissue culture cells in good agreement with their expected molecular weights (Fig. 2 D,E), indicating that these mutations do not induce degradation of Sav1 protein in mouse tissues and tissue culture cells. Therefore, a phenotype of these mice is expected to be different from that of Sav1 KO mice. Since WT, point mutant, and exon 2-deleted Sav1 expressed other known functional domains that bind to MST1/2 (Fig. 2F), and the exon 2 deletion mutant is fundamentally similar to point mutation at the Flna-binding site in terms of disruption of Flna-binding unless this domain has another biological function, we decided to further characterize these mice. Although flox/+ heterozygous mice appear to be normal presumably due to the compensation effect of WT cells and were obtained in sufficient numbers for statistical analysis, we also analyzed flox/+ heterozygous mice for their body weight, liver size, apoptosis, and histology.
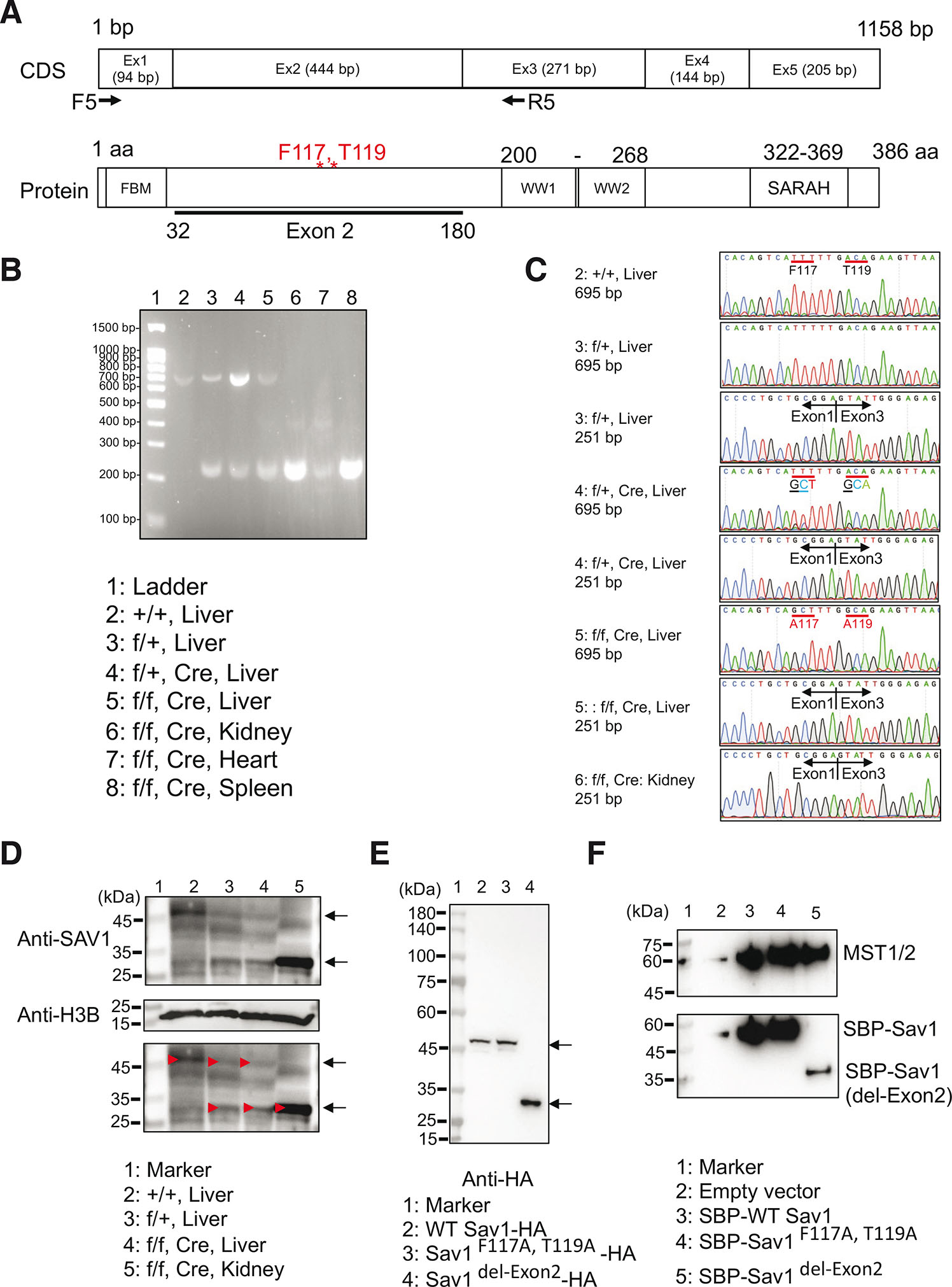
Fig. 2. Introduction of point mutations and deletion exon 2 in the Sav1 gene in the transgenic mice.
(A) Coding sequence (CDS) and protein structure of mouse Sav1. PCR using F5/R5 primers amplifies 695 bp fragment or 251 bp fragment without exon 2. Note that each exon encodes a different functional domain. (B) PCR analysis using the cDNA library from the indicated tissues. (C) Partial Sanger sequence chromatograms of the bands shown in panel F. Note that F117A/T119A mutations were successfully introduced in flox/+, Cre, and flox/flox, Cre Liver. The lower band in panel B indicates PCR product without exon 2, demonstrating that flox mouse without Cre recombinase does not express exon 2. (D) Western blotting of mouse tissues using anti-SAV1 antibodies indicated that tissues of flox/+ and flox/flox without Cre recombinase express shorter Sav1 protein. (E) Western blotting of HEK293A cells expressing HA-tagged WT, point mutant, and deletion (exon 2) mutant Sav1 using anti-HA antibodies. Arrows indicate position of WT and mutant Sav1. Calculated molecular weight of WT SAV1-HA: 46.1 kDa, Sav1F117A, T1119-HA: 46.0 kDa, Sav1del-Exon2-HA: 29.2 kDa. (F) Binding of mutant Sav1 with Mst1/2. WT SBP-Sav1, SBP-Sav1F117A, T1119, and SBP-Sav1del-Exon2 were expressed in SAV1 KO HEK293A cells and precipitated with streptavidin-coated beads. Bound proteins were detected by anti-Mst2 antibodies.
Disruption of Sav1/Flna interaction promotes apoptosis in mouse liver
To investigate the function of the Sav1-Flna interaction during mouse development, we compared the body size and liver weight of WT and transgenic mice. We found that the body weight and liver weight of homozygous mice were smaller compared to those of the same age WT and heterozygous mice (Fig. 3 A,B), whereas liver-specific Sav1 knock-out was shown to enlarge the liver (Lee et al., 2010; Lu et al., 2010). Since the normal size of Sav1 protein was detected in the liver by western blotting (Fig. 2D), growth retardation of the liver in the homozygous mice was not due to degradation of Sav1 but due to point mutation. In addition, HE staining and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay demonstrated the decreased size of the nucleus and increased apoptosis in homozygous mice livers when mice were at the age of eleven months old (Fig. 3 D-G).
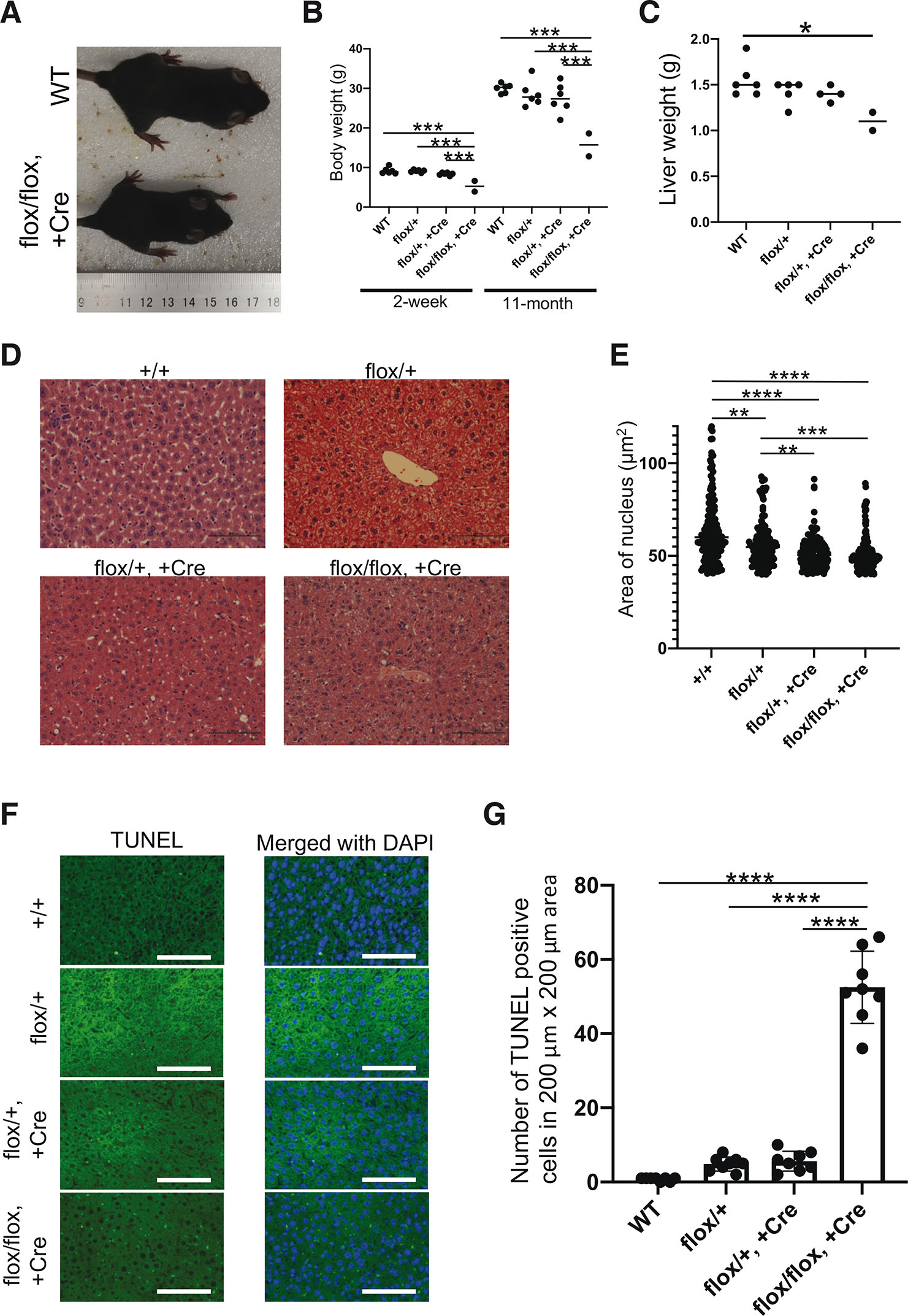
Fig. 3. Expression of non-filamin A (Flna)-binding Sav1 in mice liver promotes apoptosis and suppresses liver growth.
(A) Representative pictures of WT (+/+, top) and knock-in (KI) (flox/flox, +Cre, bottom) 5-week-old mice illustrate the smaller size of surviving KI mice. Heterozygous mice were similar to WT mice (not shown). (B) Dot plots represent the weight of 2-week-old pups and 11-month-old mice. Each dot corresponds to one mouse. The line represents the average weight of the genotype group. The body weight of the KI mice was significantly smaller than that of other mice (2-week: F(3, 16) = 17.70, P<0.0001; 11-month: F(3, 16) = 11.97, P = 0.0002). Only statistically significant p-values are shown. (C) Dot plots represent the liver weight of 11-month-old mice. Each dot corresponds to one mouse liver. The line represents the average weight of the genotype group. The liver weight of the KI mice was significantly smaller than that of other mice (F(3, 13) = 4.710, P=0.0195). Only statistically significant p-values are shown. (D) Hematoxylin & eosin (HE) staining of liver tissues from eleven-month-old mice. (E) The area of the nucleus of the HE staining (D) was measured by ImageJ and plotted. Areas smaller than 40 μm2 were omitted. n=100~200. F(3, 594) = 23.63, P<0.0001. (F) TUNEL staining of mouse livers revealed that cells underwent apoptosis. The heterozygous and homozygous sections showed increased numbers of TUNEL-positive cells. Nuclei were stained with DAPI. Scale bars: 100 μm. (G) TUNEL-positive cells were counted in 200 μm x 200 μm areas. n=8. Data are presented as mean ± SD. F(3, 28) = 181.2, P<0.0001. Significance was analyzed by one-way ANOVA followed by Tukey's multiple comparison test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Only statistically significant p-values are shown.
We also performed the HE staining and TUNEL assay for livers of two-week-old heterozygous mice. Consistent with the 11-month-old mice, the nuclear size of hepatocytes decreased (Fig. 4 A,B), and cell apoptosis was moderately increased in Cre-negative heterozygous and Cre-positive heterozygous mouse livers relative to WT controls (Fig. 4 C,D), indicating that the cell apoptosis was dominantly regulated by disruption of Sav1/Flna interaction, instead of aging of mice.
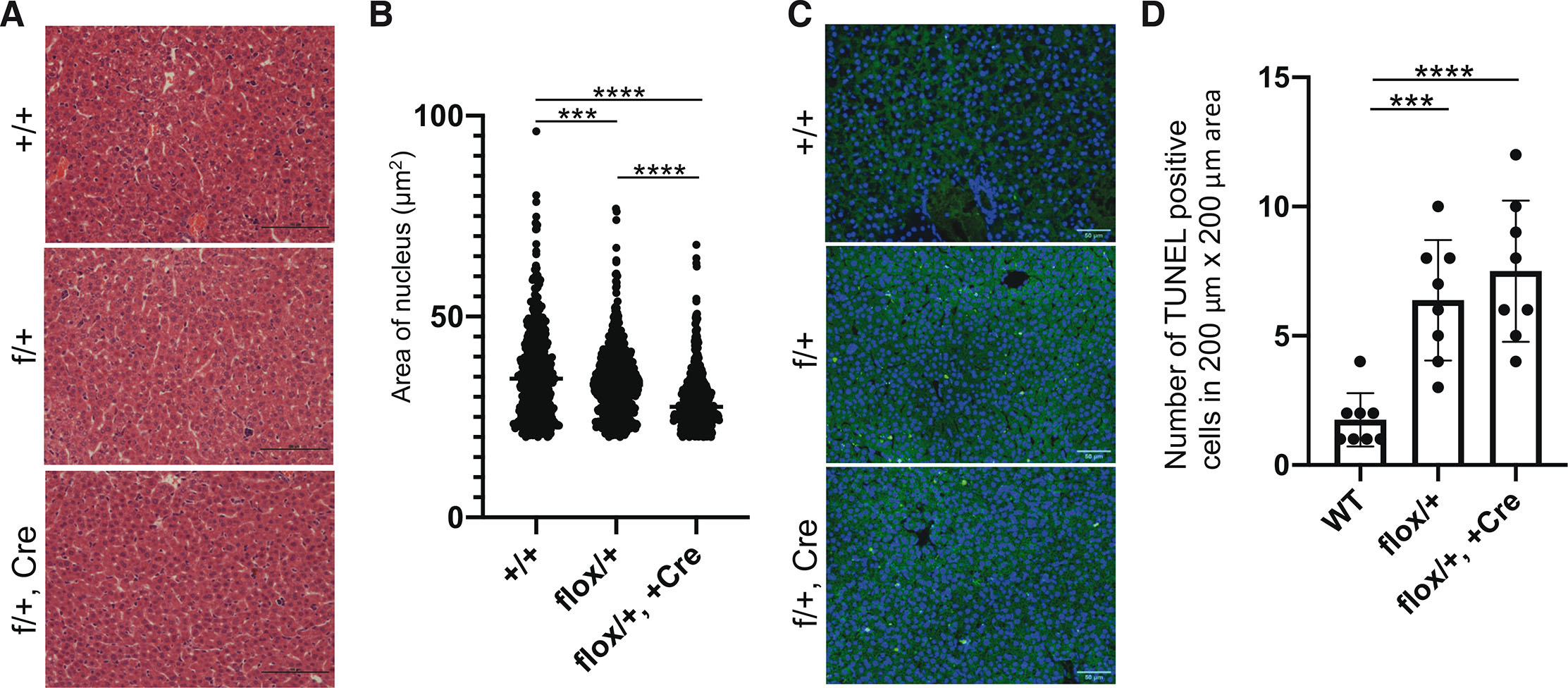
Fig. 4. Hematoxylin & eosin (HE) and TUNEL staining of 2 week-old heterozygous mouse livers.
(A) HE staining of liver sections. Scale bars: 100 μm (B) Area of the nucleus of the HE staining (A) were measured by ImageJ and plotted. Areas smaller than 40 μm2 were omitted. n=~500, F(2, 1494) = 68.38, P<0.0001. (C) TUNEL staining. Scale bars: 50 μm (D) TUNEL-positive cells were counted in 200 μm x 200 μm areas. n=8. Data are presented as mean ± SD. F(2, 21) = 16.02, P<0.0001. Significance was analyzed by one-way ANOVA followed by Tukey's multiple comparison test. ***P<0.001, ****P<0.0001. Only statistically significant p-values are shown.
Disruption of SAV1/FLNA interaction does not influence phosphorylation of YAP1
Since the disruption of the SAV1-FLNA interaction promotes translocation of YAP1 to the cytosol (Zhang et al., 2023), we determined if non-FLNA-binding mutant Sav1 induces phosphorylation of YAP1 using cells transfected with WT and mutant Sav1. However, the standard transfection protocol recommended by the manufacturer used cells at ~70-80% confluence and these cells reached confluency when cells were harvested (~24 post-transfection). Since phosphorylation of YAP1 is highly influenced by cell density (Fig. S2), we optimized the condition for transfection without reaching confluency. Consistent with the previous finding (Jiang et al., 2017), overexpression of WT Sav1 in HEK293A cells stimulated phosphorylation of YAP1 (Fig. 5). Expression of non-FLNA-binding Sav1 point and deletion mutants also increased phosphorylated YAP1 at the same extent as WT Sav1 (Fig. 5).
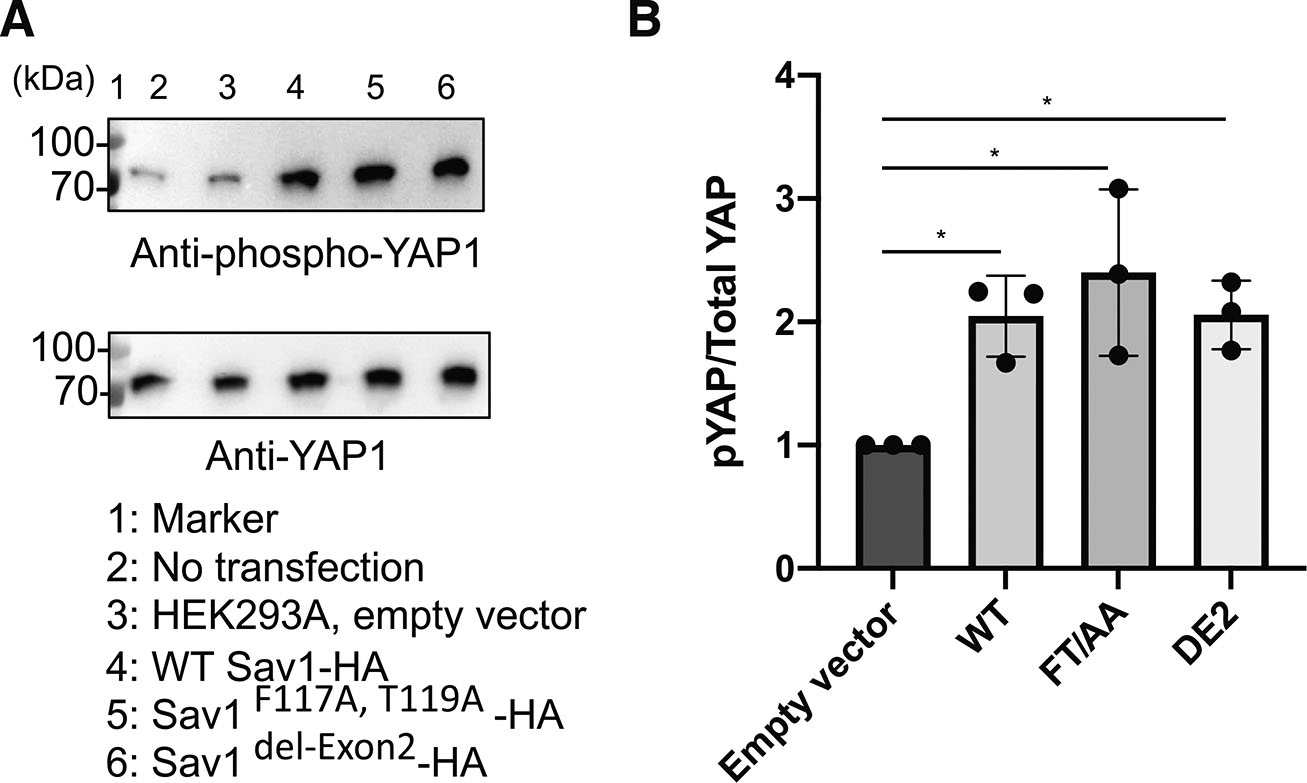
Fig. 5. Filamin A (FLNA)-binding of Sav1 does not influence phosphorylation of yes-associated protein 1 (YAP1).
(A) Western blotting showing level of YAP1 and phosphorylated YAP1 in HEK293A cells expressing WT and non-FLNA-binding mutant Sav1. (B) Quantitation of relative phosphorylation level of YAP1 in HEK293A cells expressing WT and mutant Sav1. Phosphorylated YAP1 levels were normalized to the total YAP1 levels. Significance was analyzed by one-way ANOVA followed by Newman-Keuls multiple comparisons test (*P<0.05). n=3. F(3, 8) = 6.883, P = 0.013. Data are presented as mean ± SD. Only statistically significant p-values are shown. FLNA, filamin A.
Discussion
We have recently identified SAV1 as a new FLNA-mechanobinding partner and found that disruption of this interaction releases SAV1 from FLNA in the cytoplasm and promotes diffusion of SAV1 to the nucleus and reciprocal diffusion of YAP1 from the nucleus to the cytoplasm (Zhang et al., 2023). These biochemical and cell biological data encouraged us to investigate if the force-dependent interaction of FLNA-SAV1 plays a role in animal development using transgenic mice expressing non-Flna-binding mutant Sav1. Unexpectedly, the insertion of the flox cassette skips exon 2 presumably due to its bulkiness, resulting in shorter Sav1 in all transgenic mice. Therefore, flox/flox mice express Sav1 missing exon 2 in whole bodies, and flox/flox, Cre mice express point mutant Sav1 in hepatocytes and exon 2 deletion mutant in the rest of tissues. If these mutations result in loss of function of Sav1, one would expect enlargement of the liver or tissues as liver-specific KO of Sav1 increases liver size (Lee et al., 2010; Lu et al., 2010). However, since we did not observe excessive proliferation in flox/flox and flox/flox Cre mice, point mutation or deletion of exon 2 do not lead to complete disruption of Sav1 function. Rather, proliferation was suppressed in these mice in agreement with our hypothesis, suggesting that the point and deletion mutant of Sav1 are partially functional. Although we cannot exclude the possibility that the exon 2 encoded domain has another biological function besides Flna-binding, the mutant Sav1 can at least bind to Mst kinase. Using flox/flox Cre mice and flox/+ heterozygous mice, we found that disruption of Flna-Sav1 interaction induces apoptosis, thereby retarding normal organ growth.
The FLNA-binding site is encoded by exon 2, whereas other functional domains are independently encoded by different exons. Thus, for example, the FERM binding motif (FBM) domain which binds to merlin, the PP2A A subunit, and KIBRA (Bae et al., 2017; Qi et al., 2022; Yu et al., 2010), is encoded by exon 1; the WW domains which mediate dimerization and binding to LATS (Lin et al., 2020; Tapon et al., 2002) are encoded by exon 3, and the SARAH domain which mediates heterodimerization with MST (Hwang et al., 2007) is encoded by exon 5 (Fig. 6A). Currently, function of a domain encoded by exon 4 is unknown, but this domain may provide a space to facilitate phosphorylation of LATS by MST.
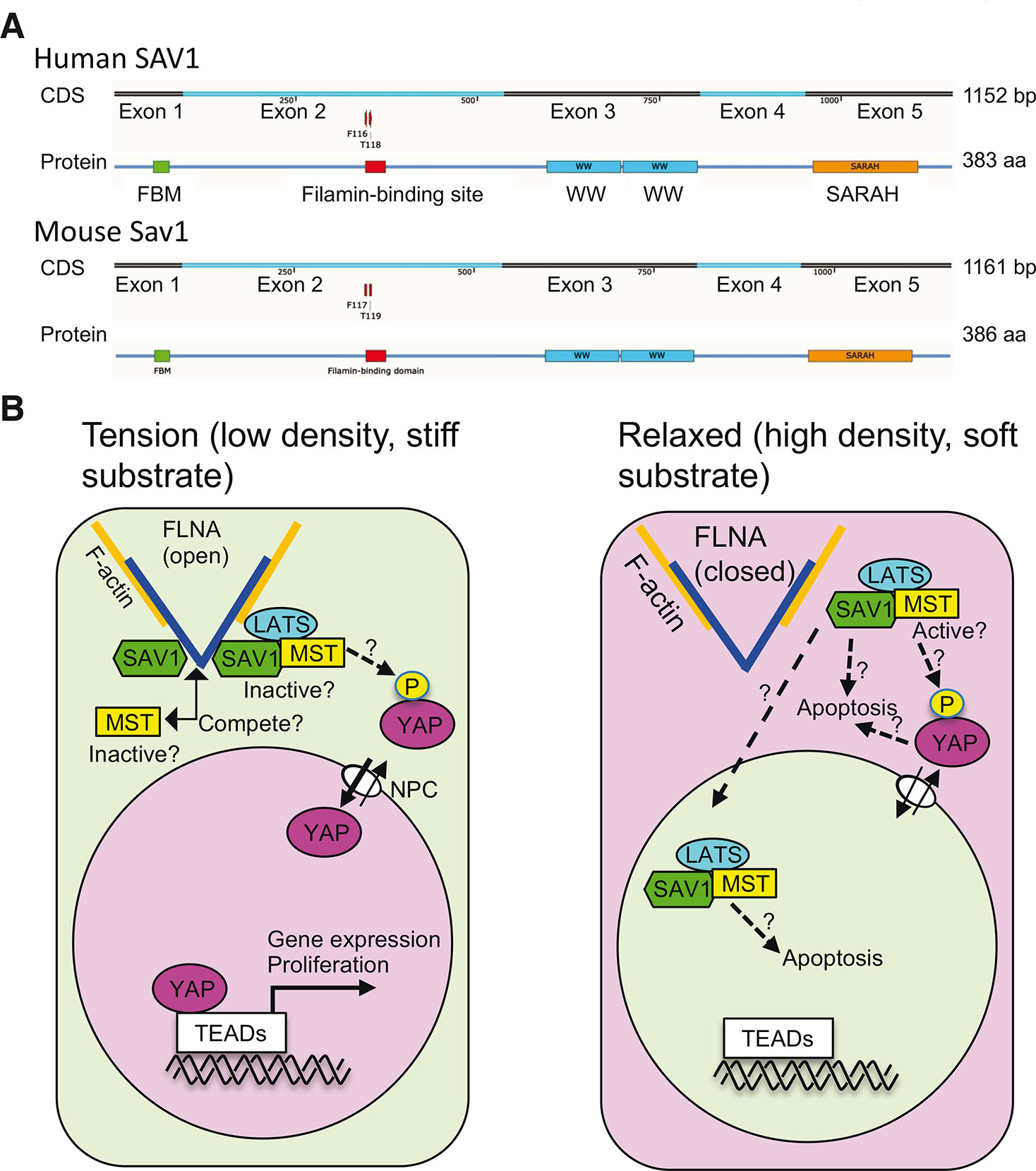
Fig. 6. How mechanical forces regulate yes-associated protein 1 (YAP1) localization through the FLNA/SAV1 interaction.
(A) Coding sequence (CDS) and protein structure of human and mouse SAV1. The FERM binding motif (FBM) (green) binds to the FERM domain of NF2, sp5-domain or WWP repeating motif (WW) domains (cyan) mediate dimerization, and the SARAH domain (orange) mediates heterodimerization with MST. (B) The binding of SAV1 to FLNA under tension retains MST1/2 and LATS1/2 in the cytoplasm, thereby endorsing the activity of YAP1 in the nucleus. In relaxed cells, SAV1 is released from FLNA to induce apoptosis by an unknown mechanism. Some of the released SAV1 might translocate to the nucleus presumably with MST1/2 and LATS1/2. Phosphorylation of YAP1 might take place in the cytosol if non-phospho-YAP1 can exit the nucleus. FLNA, filamin A; nuclear pore complex (NPC).
Insertion of mutant exon 2 gene flanked by LoxP and Lox 2272 sites resulted in deletion of exon 2 presumably due to its bulkiness, which is most likely the reason why only two homozygous mice were born among 134 littermates and these two mice had growth retardation. This result suggests that the domain encoded by exon 2 is necessary for normal growth of tissues and organs. By changing the breeding strategy, we finally obtained two Cre-positive homozygous mice that express point mutant Sav1 in hepatocytes (Sav1Hep-F116A, T118A/ Sav1Hep-F116A, T118A), while other cells express exon 2-deleted Sav1. These mice also had growth retardation similar to the homozygous F2 mice (flox/flox), indicating that mice can find an alternative pathway to survive without Flna-binding ability in about 2% chance (2 flox/flox mice out of 134 littermates and 2 homozygotes KI mice out of 90 littermates) by an unknown mechanism. TUNEL assay indicated that apoptosis was increased in the liver expressing mutant Sav1. Since we could obtain a sufficient number of heterozygous mice with and without alb-Cre expression, we histologically analyzed their livers. Consistent with the homozygous mice livers, apoptosis was increased in heterozygous mice with and without alb-Cre expression at the same extent, indicating that both point mutation of Sav1 and deletion of exon 2 induce apoptosis. However, the body sizes of heterozygous mice were normal, suggesting that WT cells can compensate for apoptotic cells induced by non-Flna-binding mutant Sav1. These results support a hypothesis that exon 2 independently encodes a domain for FLNA binding. Although SAV1 enhances MST1-mediated apoptosis through death-associated protein 4 (DAP4), histone H2B, and inhibitor of caspase-activated DNase (ICAD) (Cheung et al., 2003; Lin et al., 2002; Luo et al., 2009; Ura et al., 2001), how disruption of FLNA-SAV1 interaction induces apoptosis is an open question.
Previous research showed that mechanical stretching reduces the dimerization of Hippo (MST) and phosphorylation of Yki (YAP1), thereby promoting its nuclear localization (Fletcher et al., 2018). In addition, SAV1 is required for MST1 activation and translocation to the nucleus for subsequent LATS1/2 activation (Lee et al., 2008). Phosphorylation of YAP1 by the activated LATS1/2 excludes YAP1 from the nucleus to lead to cell apoptosis and restricts organ size overgrowth (Chen et al., 2010; Johnson and Halder, 2014; Zhang et al., 2015). Therefore, activated LATS need to enter the nucleus to phosphorylate YAP1 although it is possible that phosphorylation takes place in the cytosol if non-phospho-YAP1 can exit the nucleus (Fig. 6). Originally, we hypothesized that disruption of the FLNA-SAV1 interaction alters the phosphorylation of YAP1, but no statistical difference in the phosphorylation level of YAP1 in HEK293 cells overexpressing WT and mutant Sav1 was observed. This data suggests that apoptosis was promoted by disruption of the Flna-Sav1 interaction in mice liver not by phosphorylation of Yap1 but by an unknown mechanism. However, it is possible that phosphorylation of YAP1 reached maximum by overexpression of even WT Sav1 in the tissue culture system.
Our findings raised new questions: 1) Does the binding of SAV1 to FLNA regulate MST activity by competing with MST?, 2) Does the binding of SAV1 to FLNA reduce dimerization of MST?, 3) Does the SAV1-MST-LATS complex translocate to the nucleus to phosphorylate YAP1?, 4) How does the FLNA-SAV1 interaction regulate apoptosis (Fig. 6B).
In conclusion, using transgenic mice expression non-Flna-binding Sav1 with either point mutations or deletion of exon 2, we show that force-dependent FLNA-SAV1 interaction prevents apoptosis with yet unknown mechanisms to control the growth of tissues and organs. Contrary to the widely recognized paradigms, depletion of YAP/TAZ (WW domain-containing transcription regulator protein 1) in mouse liver resulted in larger liver size compared to wild-type counterparts, although liver regeneration is less efficient when YAP/TAZ are deleted (Lu et al., 2018; Verboven et al., 2021). This suggests that YAP/TAZ are not essential for liver growth, but potent inducers of cell proliferation which is controlled by the Hippo pathway (Russell and Camargo, 2022). Therefore, one can hypothesize that loss of YAP/TAZ led to the enlargement of the liver because the force-dependent FLNA-SAV1 interaction lost its effectors to control YAP/TAZ activity. To further test this possibility using an animal model, the Flna-Sav1 interaction needs to be specifically targeted without affecting other functions.
Materials and Methods
Antibodies
Rabbit anti-YAP antibody was purchased from Cell Signaling Technology (#4912S). Anti-phospho-YAP (Ser127) antibody was purchased from Cell Signaling Technology (#4911). A mouse monoclonal anti-HA antibody was purchased from Proteintech (#66006-2-Ig). Rabbit polyclonal anti-Histone H3 antibody was purchased from Beyotime (#AH433-1). Rabbit monoclonal anti-STK3/MST-2 antibody was purchased from Abcam (ab52641). A mouse monoclonal anti-SAV1 (3B2) antibody was purchased from Novus Biologicals. Rabbit polyclonal anti-SAV1 antibodies were generated as follows: His-SAV1 (130-383 aa) protein was expressed in BL21 (DE3) using pET-23a (+)-HTb vector, purified using Ni-NTA affinity resin (Qiagen) in the presence of 8 M urea, and immunized to rabbit (outsourced to Pacific immunology). Antibodies were affinity purified using GST-SAV1 (199-262aa) immobilized on NHS-Sepharose. For western blotting, the purified antibodies were used at 0.66 μg/ml. Goat anti-Rabbit (H+L) IgG-HRP conjugate secondary antibody (#172-1019) and goat anti-Mouse IgG (H+L)-HRP conjugate (#172-1011) secondary antibody were purchased from BioRAD.
Animal care and treatment
This study complies with all relevant ethical regulations for which approval of the institutional review board and the ethics committee of Tianjin University. All animal care and procedures were carried out according to the approval of the Institute of Radiation Medicine Chinese Academy of Medical Sciences. Mice were housed at barrier condition using a 12 h light to 12 h dark cycle and following Laboratory Rodent Diet 5001 (PMI International). Mice were sacrificed by the mouse spinal cord dislocation method. The extracted organs were frozen in liquid nitrogen or immersed in fixatives (Servicebio).
Construction of chimeric Sav1 (F117A/T119A, +) knock-in mouse
To generate the mutant Sav1 knock-in ES cells (Fig. 1A), the mutant exon 2 was cloned into intron 2 in the reverse orientation in the targeting vector. The WT and mutant exon 2 were flanked with loxP and lox2272 sites, and the Neo cassette was flanked by self-deletion anchor sites. F117A (TTT to GCT) and T119A (ACA to GCA) mutations (corresponding to F116A and T118A mutations of human SAV1 that disrupt FLNA interaction (Zhang et al., 2023).) were introduced into the mutant exon 2. Diphtheria toxin A was used for negative selection. The heterozygous chimeric Sav1(F117A/T119A, +) mice were generated from Cyagen Biosciences Inc. Targeted ES cell clone (Fig. 1A) was injected into C57BL/6 albino embryos, which were re-implanted into CD-1 pseudo-pregnant female mice. Founder animals were identified by their coat color, and their germline transmission was confirmed by breeding with C57BL/6 females and subsequent genotyping of the offspring. The Neo cassette is self-deleted in germ cells so that the offspring were Neo cassette-free. Positive pups from the ES clone were identified as positive by PCR screening for loxP, lox2272, and Neo deletion (Fig. S1). Primer for loxP: loxP-F (F1): 5’-CATTCAAATCAGCAGCCATGACC-3’; loxP-R (R1): 5’-ACCCGGTTCTAAGCACAATTATCAA-3’. Primers for lox2272 PCR:
lox2272-F (F2): 5’-ACTGCGCCCTAATAACTTCGTA-3’; lox2272-R (R2): 5’-GTGTATGCTGGTTTTGTCTCACTAA-3’. Primers for Neo deletion: Neo-del-F (F3): 5’-CGAGGTGGGGATTAAGGAATGTT-3’; Neo-del-R (R3): 5’-GTCACGTGGTAGCTCGATATAACT-3’. Primers for constitutive KI allele: cKI-F (F4): 5’-GGAATGTTGCAGCTCCACTTAAT-3’; cKI-R (R4): 5’-TCCCAAGAGGATCAAGAAAATGCTA-3’. Primers for Cre transgene: Forward1: 5’-CATATTGGCAGAACGAAAACGC-3’ Reverse1: 5’-CCTGTTTCACTATCCAGGTTACGG-3’.
TUNEL assay
Liver sections were deparaffinized and rehydrated followed by digestion with proteinase K (22 min at 37°C), and then permeabilized by permeabilizing working solution for 20 min at room temperature. After incubating with the TUNEL reaction mixture for 2 h, sections were sealed with an antifade mounting medium. Adherent cells were centrifuged (200 x g, 5 min), perfused with 4% formaldehyde in PBS for 30 min, rinsed with PBS, permeabilized by 0.3% Triton X-100 in PBS for 5 min, and incubated with TUNEL reaction mixture (Beyotime) for 1hr. Positive controls were treated with DNase I (TAKARA) for 20 min before incubating with a TUNEL reaction mixture. The TUNEL-positive cells were observed and imaged under a fluorescence microscope.
Reverse Transcription PCR (RT-PCR)
Total RNA was extracted according to the instruction of RNAiso Plus kit (#9108, TAKARA). Total mRNA was reversely transcribed into the cDNA library by First-Strand cDNA Synthesis kit (#N20909, Transgen). PCR amplification was performed using 5’ primer (F5), 5’-GAGGTGTCTAAGCCGGCC-3’, and 3’ primer (R5) 5’-CAGCCAGGAGGAAGTCCTTCTC-3’, and PCR product was analyzed on agarose gel and sequenced.
Histochemistry
Tissue sections were deparaffinized, rehydrated, and then subjected to antigen retrieval by heating in citric acid (pH 6.0). After incubating with 3% hydrogen peroxide for 25 min at room temperature, tissue sections were blocked with 3% BSA for 30min at room temperature, in turn, incubated with primary antibodies prepared with PBS (pH 7.4) overnight at 4˚C and secondary antibodies (HRP-labeled) for 50 min at room temperature. Then the specimen was treated with a freshly prepared DAB developing solution. The sections were counterstained with hematoxylin stain solution and treated with hematoxylin returning blue solution. The sections were dehydrated, removed from xylene, and mounted with neutral gum. Tissue sections were visualized under fluorescence microscopy (Nikon ECLIPSE 80i).
Plasmid construction
Mouse Sav1 (wild-type (WT), F117A/T119A, and deletion exon 2) was amplified by PCR using 5’ primer, 5’-GTACCGGATCCATGCTGTCCCGCAAGAAAACC-3’, and 3’primer, 5’-GATATGCGGCCGCACTTAAGAACGTCTTGCCATGCTG-3’, using cDNA library from WT and KI mice as a template, digested with BamHI/NotI, and ligated into pcDNA3.6-HA vector at BamHI/NotI sites. pSBP (streptavidin-binding protein, GHVVEGLAGELEQLRARLEHHPQGQ)-C1 was constructed by ligating annealed double strand DNA of Age I-SBP (5’-CCGGTATGGGCCACGTGGTGGAGGGCCTGGCCGGCGAGCTGGAGCAGCTGAGAGCCAGACTGGAGCACCACCCCCAGGGCCAGAGAGAGG-3’) and SBP-BamH I (5’-GATCCCTCTCTCTGGCCCTGGGGGTGGTGCTCCAGTCTGGCTCTCAGCTGCTCCAGCTCGCCGGCCAGGCCCTCCACCACGTGGCCCATA-3’) into pFLAG-C1 vector digested with Age I/BamH I. pSBP-Sav1 was constructed by amplifying mouse Sav1 with 5’ primer, 5’-GTACCGGATCCATGCTGTCCCGCAAGAAAACC-3’ and 3’ primer, 5’-GGGCGCGGCCGCTTAACTTAAGAACGTCTTGCCATGCTG-3’ using pcDNA3 Sav1(WT, FT/AA, and Del exon 2)-HA as templates, digesting with BamHI/NotI and ligating into pSBP vector at BamHI and NotI sites. SAV1 KO plasmid was constructed as follows. The guide RNAs (gRNAs) were designed using the design tool at https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design. The sgRNAs (F1 sense: 5’-CACCGGAAGTCCTTCTCGCTCAAG-3’ and R1 antisense: 5’-AAACCTTGAGCGAGAAGGACTTCC-3’; F2 sense: 5’-CACCGCTGTGGACTGGACAATGAG-3’ and R2 antisense: 5’-AAACCTCATTGTCCAGTCCACAGC-3’) were annealed in the tube containing 7 µl ultrapure H2O, 1 µl 10 × annealing buffer (Tris-HCl 100 mM, NaCl 1 M, EDTA (pH 8.0) 0.1 mM), 1 µl of 50 µM sense oligo and 1 µl of 50 µM antisense oligo followed the program 95 °C, 5 min ->25 °C (1 °C/min), respectively. The annealed oligonucleotides were purified with PCR-Pure Kit (Omega). pX330-U6-Chimeric_BB-CBh-hSpCas9 plasmid (Addgene) was digested with BbsI-HF (New England Biolabs) and ligated with the gRNA.
Cell culture and transfection
HEK293A cells were cultured in DMEM high glucose with 10% fetal bovine serum and penicillin-streptomycin. Cells were maintained at 37°C and 5% CO2. Before transfection, 0.5~1x105 cells were plated on a 6-well plate so that the cell confluency will be 30~60% after 24 hours. The medium was changed to a fresh growth medium without antibiotics at the time of transfection. Plasmids (4 µg) were mixed with 10 µl of LipoGeneTM 2000 Star Transfection Reagent (US Everbright) in 500 µl Opti-MEM® I Reduced Serum Medium (Gibco) and incubated for 20 min at room temperature. The mixture was drop-wisely added into each well and then incubated for another 24 hours.
SAV1 KO HEK293 cells
HEK293A cells were cultured on a 24-well plate at 70% confluence at the time of transfection. pX330-Sav1 gRNA (1 µg) was transfected with 2 µl of LipoGeneTM 2000 Star Transfection Reagent (US Everbright). After 48 hours, the cells were seeded on 96-well plates by serial dilution. Clones were detectable by microscopy after one week. These colonies were subcultured into 12-well plates. When the cells reached 80-90% confluency, cells were lysed by ice-cold RIPA buffer (Sparkjade) with an EDTA-free protease inhibitor cocktail (Sigma) at 1:100 just before lysis. Cell lysis was solubilized by 5 × SDS sample buffer (10% SDS, 50% glycerol, 250 mM Tris-HCl (pH6.8), 5 mM EDTA, 0.25% (w/v) bromophenol blue dye, 25% beta-mercaptoethanol. Western blotting was performed to confirm the absence of SAV1 with anti-SAV1 monoclonal antibodies.
Western blotting
Mouse tissue was lysed by ice cold RIPA (Sparkjade) and 1% (v/v) protease inhibitor (Sigma) and homogenized with a glass homogenizer, then maintained for 40 min on ice. Then samples were centrifuged for 20 min at 13,400 × g at 4 °C. Total protein concentration was estimated by BCA assay kit (Meilunbio). Tissue culture cells were rinsed with PBS, lysed with 1x SDS sample buffer, sonicated for 5 seconds, then boiled at 95˚C for 5 min. The sample was loaded on SDS-PAGE, and proteins were transferred onto 0.45 µm nitrocellulose membrane. The membrane was blocked with blocking buffer (5% non-fat milk in TBST (20 mM Tris-HCl, pH 7.4, 110 mM NaCl, 5 mM MgCl2, 0.1% Tween 20). Primary antibodies were prepared in the blocking solution and membranes were incubated overnight at 4°C. The membrane was washed with TBST and incubated with HRP-conjugated secondary antibodies in TBST for 1 h at room temperature. The membrane was washed and developed with the HRP substrate (WesternBright ECL, Advansta).
Quantitation of phosphorylated YAP1 in HEK293A cells expressing WT and mutant Sav1
Band images of western blotting of anti-YAP and anti-phospho YAP were imported into Image J (version 1.48) and band intensity was calculated as follows. Images were transformed into 8-bit. After subtracting the background, the images were inverted. The bands were individually selected and band intensity was measured. The data was imported into GraphPad Prism version 5.00 (La Jolla, CA) for analysis.
Pull-down assay with SBP-Sav1
SBP-Sav1 was expressed in SAV1-KO HEK293A cells on a 60 mm dish by transfecting 8 µg plasmids using LipoGeneTM 2000 Star transfection reagent (US Everbright). HEK293A cells expressing SBP-Sav1 were lysed with lysis buffer (50 mM Tris-HCl (pH7.4), 150 mM NaCl, 0.1 mM β-mercaptoethanol, 1 mM EGTA, 1% TritonX-100). Protease inhibitor (Beyotime) and 0.5 M EDTA (Beyotime) were added just before lysing at the ratio of 1:100. The cell lysates were centrifuged at 15,000 x g for 10 min at 4˚C. The supernatant was collected and incubated with streptavidin (Genscript) coated NHS-activated Sepharose (Cytiva) at 4˚C for 4 hours. The streptavidin beads were washed with washing buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.1 mM β-mercaptoethanol, 1 mM EGTA, 0.1% TritonX-100) three times. Bound proteins were solubilized in SDS sample buffer, separated on SDS-PAGE, and transferred to a nitrocellulose membrane. Western blotting was performed using anti-STK3/MST-2 (ab52641, Abcam), anti-LATS1 (C66B5, Cell Signaling Technology), anti-SAV1 (3B2, Novus Biologicals) and anti-beta actin (N10218, Transgene) antibodies. After washing, the membrane was incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. The membrane was washed and developed with the HRP substrate (WesternBright ECL, Advansta).
Statistics
Data are mean ± S.D. All experiments were performed at least three times independently. All image analysis was performed by operators who were blinded to the treatments administered. Significance was analyzed by one-way ANOVA followed by Tukey's multiple comparison test or Newman-Keuls multiple comparisons test. * P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Only statistically significant p-values are shown. Statistical analysis was performed in GraphPad Prism (version 9.0.0, GraphPad Software Inc., San Diego, CA, USA).
Supplementary Material
Acknowledgements
This work was supported by grants from the Natural Science Foundation of China to F.N. (grant number 32070777).
Abbreviations
delExon2 or DE2, deletion exon 2 mutation ; ES, embryonic stem ; FBM, FERM binding motif ; FT/AA, F117A and T119A mutation in mouse Sav1 ; HE staining, hematoxylin and eosin staining ; FLNA, filamin A ; KI, knock-in ; NPC, nuclear pore complex ; SAV1, Protein salvador homolog 1 ; TAZ, WW domain-containing transcription regulator protein 1 ; TUNEL assay, terminal deoxynucleotidyl transferase dUTP nick-end labeling assay ; WT, wild-type ; WW, rsp5-domain or WWP repeating motif ; YAP-1, yes-associated protein 1 ;References
Bae S. J., Ni L., Osinski A., Tomchick D. R., Brautigam C.A., Luo X., (2017). SAV1 promotes Hippo kinase activation through antagonizing the PP2A phosphatase STRIPAK. Elife 6: 30278.
Callus B. A., Verhagen A. M., Vaux D. L. (2006). Association of mammalian sterile twenty kinases, Mst1 and Mst2, with hSalvador via C-terminal coiled-coil domains, leads to its stabilization and phosphorylation. FEBS Journal 273: 4264-4276.
Chen L., Loh P. G., Song H. (2010). Structural and functional insights into the TEAD-YAP complex in the Hippo signaling pathway. Protein & Cell 1: 1073-1083.
Cheng Y., Mao M., Lu Y. (2022). The biology of YAP in programmed cell death. Biomarker Research 10: 34.
Cheung W. L., Ajiro K., Samejima K., Kloc M., Cheung P., Mizzen C. A., Beeser A., Etkin L. D., Chernoff J., Earnshaw W. C., Allis C.D. (2003). Apoptotic Phosphorylation of Histone H2B Is Mediated by Mammalian Sterile Twenty Kinase. Cell 113: 507-517.
Das A., Fischer R. S., Pan D., Waterman C. M. (2016). YAP Nuclear Localization in the Absence of Cell-Cell Contact Is Mediated by a Filamentous Actin-dependent, Myosin II- and Phospho-YAP-independent Pathway during Extracellular Matrix Mechanosensing. Journal of Biological Chemistry 291: 6096-6110.
de Amorim S. S., de Sousa Rodrigues M. M., Mencalha A. L. (2021). The tumor suppressor role of salvador family WW domain-containing protein 1 (SAV1): one of the key pieces of the tumor puzzle. Journal of Cancer Research and Clinical Oncology 147: 1287-1297.
Dupont S., Morsut L., Aragona M., Enzo E., Giulitti S., Cordenonsi M., Zanconato F., Le Digabel J., Forcato M., Bicciato S., Elvassore N., Piccolo S. (2011). Role of YAP/TAZ in mechanotransduction. Nature 474: 179-183.
Elosegui-Artola A., Andreu I., Beedle A. E.M., Lezamiz A., Uroz M., Kosmalska A. J., Oria R., Kechagia J. Z., Rico-Lastres P., Le Roux A.L., Shanahan C. M., Trepat X., Navajas D., Garcia-Manyes S., Roca-Cusachs P. (2017). Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 171: 1397-1410.e14.
Finch-Edmondson M., Sudol M. (2016). Framework to function: mechanosensitive regulators of gene transcription. Cellular & Molecular Biology Letters 21: 28.
Fletcher G. C., Diaz-de-la-Loza M.C., Borreguero-Muñoz N., Holder M., Aguilar-Aragon M., Thompson B. J. (2018). Mechanical strain regulates the Hippo pathway in Drosophila . Development 145: dev159467.
Hwang E., Ryu K.S., Pääkkönen K., Güntert P., Cheong H.K., Lim D.S., Lee J.O., Jeon Y. H., Cheong C. (2007). Structural insight into dimeric interaction of the SARAH domains from Mst1 and RASSF family proteins in the apoptosis pathway. Proceedings of the National Academy of Sciences 104: 9236-9241.
Jiang J., Chang W., Fu Y., Gao Y., Zhao C., Zhang X., Zhang S. (2017). SAV1 represses the development of human colorectal cancer by regulating the Akt-mTOR pathway in a YAP-dependent manner. Cell Proliferation 50: e12351.
Johnson R., Halder G. (2014). The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nature Reviews Drug Discovery 13: 63-79.
Kassianidou E., Kalita J., Lim R. Y.H. (2019). The role of nucleocytoplasmic transport in mechanotransduction. Experimental Cell Research 377: 86-93.
Lee J.H., Kim T.S., Yang T.H., Koo B.K., Oh S.P., Lee K.P., Oh H.J., Lee S.H., Kong Y.Y., Kim J.M., Lim D.S. (2008). A crucial role of WW45 in developing epithelial tissues in the mouse. The EMBO Journal 27: 1231-1242.
Lee K.P., Lee J.H., Kim T.S., Kim T.H., Park H.D., Byun J.S., Kim M.C., Jeong W.I., Calvisi D. F., Kim J.M., Lim D.S. (2010). The Hippo–Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proceedings of the National Academy of Sciences 107: 8248-8253.
Leung J. Y., Wilson H. L., Voltzke K. J., Williams L. A., Lee H. J., Wobker S. E., Kim W. Y. (2017). Sav1 Loss Induces Senescence and Stat3 Activation Coinciding with Tubulointerstitial Fibrosis . Molecular and Cellular Biology 37: e00565-16..
Lin Y., Khokhlatchev A., Figeys D., Avruch J. (2002). Death-associated Protein 4 Binds MST1 and Augments MST1-induced Apoptosis. Journal of Biological Chemistry 277: 47991-48001.
Lin Z., Xie R., Guan K., Zhang M. (2020). A WW Tandem-Mediated Dimerization Mode of SAV1 Essential for Hippo Signaling. Cell Reports 32: 108118.
Lu L., Finegold M. J., Johnson R. L. (2018). Hippo pathway coactivators Yap and Taz are required to coordinate mammalian liver regeneration. Experimental & Molecular Medicine 50: e423-e423.
Lu L., Li Y., Kim S. M., Bossuyt W., Liu P., Qiu Q., Wang Y., Halder G., Finegold M. J., Lee J.S., Johnson R. L. (2010). Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proceedings of the National Academy of Sciences 107: 1437-1442.
Luo X., Li Z., Yan Q., Li X., Tao D., Wang J., Leng Y., Gardner K., Judge S. I., Li Q. Q., Hu J., Gong J. (2009). The human WW45 protein enhances MST1-mediated apoptosis in vivo. International Journal of Molecular Medicine 23: 357-362.
Maehama T., Nishio M., Otani J., Mak T. W., Suzuki A. (2021). The role of Hippo‐YAP signaling in squamous cell carcinomas. Cancer Science 112: 51-60.
Oh H., Irvine K. D. (2008). In vivo regulation of Yorkie phosphorylation and localization. Development 135: 1081-1088.
Piccolo S., Dupont S., Cordenonsi M. (2014). The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiological Reviews 94: 1287-1312.
Qi S., Zhu Y., Liu X., Li P., Wang Y., Zeng Y., Yu A., Wang Y., Sha Z., Zhong Z., Zhu R., Yuan H., Ye D., Huang S., Ling C., Xu Y., Zhou D., Zhang L., Yu F.X. (2022). WWC proteins mediate LATS1/2 activation by Hippo kinases and imply a tumor suppression strategy. Molecular Cell 82: 1850-1864.e7.
Racanelli V., Rehermann B. (2006). The liver as an immunological organ. Hepatology 43: S54-S62.
Russell J. O., Camargo F. D. (2022). Hippo signalling in the liver: role in development, regeneration and disease. Nature Reviews Gastroenterology & Hepatology 19: 297-312.
Seo E., Kim W.Y., Hur J., Kim H., Nam S. A., Choi A., Kim Y.M., Park S. H., Chung C., Kim J., Min S., Myung S.J., Lim D.S., Kim Y. K. (2016). The Hippo-Salvador signaling pathway regulates renal tubulointerstitial fibrosis. Scientific Reports 6: 31931.
Taniguchi K., Wu L.W., Grivennikov S. I., de Jong P. R., Lian I., Yu F.X., Wang K., Ho S. B., Boland B. S., Chang J. T., Sandborn W. J., Hardiman G., Raz E., Maehara Y., Yoshimura A., Zucman-Rossi J., Guan K.L., Karin M. (2015). A gp130–Src–YAP module links inflammation to epithelial regeneration. Nature 519: 57-62.
Tapon N., Harvey K. F., Bell D. W., Wahrer D. C.R., Schiripo T. A., Haber D. A., Hariharan I. K. (2002). salvador Promotes Both Cell Cycle Exit and Apoptosis in Drosophila and Is Mutated in Human Cancer Cell Lines. Cell 110: 467-478.
Tran T., Mitra J., Ha T., Kavran J. M. (2020). Increasing kinase domain proximity promotes MST2 autophosphorylation during Hippo signaling. Journal of Biological Chemistry 295: 16166-16179.
Ura S., Masuyama N., Graves J. D., Gotoh Y. (2001). MST1-JNK promotes apoptosis via caspase-dependent and independent pathways. Genes to Cells 6: 519-530.
Verboven E., Moya I. M., Sansores-Garcia L., Xie J., Hillen H., Kowalczyk W., Vella G., Verhulst S., Castaldo S. A., Algueró-Nadal A., Romanelli L., Mercader-Celma C., Souza N. A., Soheily S., Van Huffel L., Van Brussel T., Lambrechts D., Roskams T., Lemaigre F. P., Bergers G., van Grunsven L. A., Halder G. (2021). Regeneration Defects in Yap and Taz Mutant Mouse Livers Are Caused by Bile Duct Disruption and Cholestasis. Gastroenterology 160: 847-862.
Yao F., Zhou Z., Kim J., Hang Q., Xiao Z., Ton B. N., Chang L., Liu N., Zeng L., Wang W., Wang Y., Zhang P., Hu X., Su X., Liang H., Sun Y., Ma L. (2018). SKP2- and OTUD1-regulated non-proteolytic ubiquitination of YAP promotes YAP nuclear localization and activity. Nature Communications 9: 2269.
Yin F., Yu J., Zheng Y., Chen Q., Zhang N., Pan D. (2013). Spatial Organization of Hippo Signaling at the Plasma Membrane Mediated by the Tumor Suppressor Merlin/NF2. Cell 154: 1342-1355.
Yu J., Zheng Y., Dong J., Klusza S., Deng W.M., Pan D. (2010). Kibra Functions as a Tumor Suppressor Protein that Regulates Hippo Signaling in Conjunction with Merlin and Expanded. Developmental Cell 18: 288-299.
Zeng Q., Hong W. (2008). The Emerging Role of the Hippo Pathway in Cell Contact Inhibition, Organ Size Control, and Cancer Development in Mammals. Cancer Cell 13: 188-192.
Zeybek N. D., Baysal E., Bozdemir O., Buber E. (2021). Hippo Signaling: A Stress Response Pathway in Stem Cells. Current Stem Cell Research & Therapy 16: 824-839.
Zhang H., Mao Z., Yang Z., Nakamura F. (2023). Identification of Filamin A Mechanobinding Partner III: SAV1 Specifically Interacts with Filamin A Mechanosensitive Domain 21. Biochemistry 62: 1197-1208.
Zhang K., Qi H.X., Hu Z.M., Chang Y.N., Shi Z.M., Han X.H., Han Y.W., Zhang R.X., Zhang Z., Chen T., Hong W. (2015). YAP and TAZ Take Center Stage in Cancer. Biochemistry 54: 6555-6566.
Zhao B., Lei Q.Y., Guan K.L. (2008). The Hippo–YAP pathway: new connections between regulation of organ size and cancer. Current Opinion in Cell Biology 20: 638-646.
Zhou W., Zhao M. (2018). How Hippo Signaling Pathway Modulates Cardiovascular Development and Diseases. Journal of Immunology Research 2018: 1-8.
Zinatizadeh M. R., Miri S. R., Zarandi P. K., Chalbatani G. M., Rapôso C., Mirzaei H. R., Akbari M. E., Mahmoodzadeh H. (2021). The Hippo Tumor Suppressor Pathway (YAP/TAZ/TEAD/MST/LATS) and EGFR-RAS-RAF-MEK in cancer metastasis. Genes & Diseases 8: 48-60.