Int. J. Dev. Biol. 66: 77 - 83 (2022)
Special Issue: Developmental Biology in Greece
Vascular Endothelial (VE)-cadherin-mediated adherens junctions involvement in cardiovascular progenitor cell specification
Open Access | Review | Published: 26 October 2021
Abstract
Vascular Endothelial cadherin, a type II classical cadherin, is the major cadherin molecule participating in homotypic cell-cell adhesion structures between endothelial cells. It associates with cytoplasmic and membrane cytoskeletal elements to form endothelial adherens junctions (AJs), pivotal in regulating endothelial barrier function in the adult. VE-cadherin-mediated AJs are also involved in signaling via direct or indirect associations with receptors. The generation of mutant animals, especially mice and zebrafish, revealed many details concerning the role of VE-cadherin-mediated AJs in cardiovascular development. In general, VE-cadherin knockout (KO) in mice is embryonic lethal due to severe cardiovascular defects, and major signaling pathways as well as vascular formation cues were discovered in developing endothelium. However, there is little information regarding AJs formation and their components in cardiovascular progenitors. We have characterized in detail the activation pattern of mouse VE-cadherin promoter (Pvec) in a mouse embryonic stem cells (ESCs) differentiation system in vitro. Surprisingly, we found that it is activated transiently in cardiac progenitors that belong to the second heart field. Based on Pvec activation, we isolated this population in vitro and found that it can self-renew by induction of the Wnt/β-catenin pathway. Next, we successfully established cell culture conditions that allowed self-renewal of this population that consists of endothelial and cardiac progenitors. Transplantation in rat hearts showed that they can survive and differentiate to cardiomyocytes and endothelial cells. Although further characterization is needed, these cells can be used in cell-based therapies as well as in drug screening.
Keywords
adherens junctions, cardiovascular progenitors, VE-cadherin
Adherens junctions (AJs) are cell-cell adhesion structures pivotal to morphogenetic processes, lineage specification and proliferation. They provide adhesive properties through classical cadherin receptors and the cytoplasmic catenins to support forming tissues, and are also involved in segregation of various cell-types emerging during differentiation (Harris and Tepass, 2010). Classical cadherins are Ca+2-dependent transmembrane molecules responsible for cell-cell interactions, and constitute the backbone of AJs defined by the presence of five repetitive homologous “cadherin domains” in their extracellular domains. Classical cadherins associate with catenins through their cytoplasmic domains. While the extracellular N-terminal cadherin domains engage in homophilic interactions, their C-terminal cytoplasmic domains interact with β-catenin, plakoglobin and p120, which couple the actin or intermediate filament cytoskeleton to AJs.
Cadherins functions are not limited to “cellular glue” but are also involved directly or indirectly with crucial signaling pathways such as tyrosine receptor kinase, and can regulate their activities (Halbleib and Nelson, 2006; Stepniak et al., 2009; Niessen et al., 2011; Klezovitch and Vasioukhin, 2015). Differentiating cells are equipped with a repertoire of cadherins that determine their associations between similar or different cell-types emerging during embryogenesis (Hulpiau and van Roy, 2009). E-cadherin is the first cadherin expressed during preimplantation developmental stages in trophoectoderm, as well as in emerging pluripotent epiblast and extraembryonic primitive endoderm cells. Cadherin-switch is a major developmental process occurring during lineage specification (Wheelock et al., 2008). Most significantly, cadherin switch takes place during gastrulation when epiblast cells undergo epithelial to mesenchymal transition to form mesodermal cells expressing N-cadherin (Acloque et al., 2009).
N- and VE-cadherin are the major cadherins expressed in endothelial cells. However, VE-cadherin is specific to endothelial AJs. N-cadherin was shown to be essential for cardiovascular system formation, and VE-cadherin expression is regulated by N-cadherin during early embryogenesis. Loss of N-cadherin in endothelial cells results in embryonic lethality at mid-gestation due to major disruption of vascular structures, and caused decrease in VE-cadherin expression (Luo and Radice, 2005).
VE-cadherin is localized at the endothelial cell-cell junctions, where it mediates cell adhesion and plays a crucial role in vasculogenesis and angiogenesis. It is well established that VE-cadherin plays a central role in endothelial cell-cell adhesion regulation and vascular endothelial barrier function in the adult (Komarova et al., 2017). In addition, is involved in many aspects of endothelial cell biology, such as cell proliferation, survival, cell shape, cell migration and cell polarity (Iden et al., 2006). Formation of junctional mechanosensory complexes consisting of VE-cadherin, PECAM-1 and vascular-endothelial growth factor receptor 2 (VEGFR2) sense and transmit mechanical forces that modulate signaling pathways described in the adult endothelial cells (Tzima et al., 2005; Harris and Tepass, 2010; Conway et al., 2013; Dejana and Vestweber, 2013).
VE-cadherin in signaling pathways during cardiovascular system development
VE-cadherin involvement in signaling has been previously reported. The VE-cadherin association with VEGFR2 results in mitogen-activated protein kinase (MAPK) negative regulation (Grazia Lampugnani et al., 2003; Lampugnani et al., 2006). On the other hand, it positively regulates PI3-kinase (PI3K)/Akt pathway, through β-catenin, thus promoting anti-apoptotic signaling (Carmeliet et al., 1999). VE-cadherin association with the transforming growth factor-beta (TGF-β) receptor complex regulates TGF-β proliferation signaling (Rudini et al., 2008). In addition, VE-cadherin mediated AJs regulate the subcellular localization of YAP through VE-cadherin/PI3K/Akt activation, resulting in YAP phosphorylation and its inactivation in endothelial cells (Choi and Kwon, 2015; Giampietro et al., 2015). VE-cadherin was found to associate with ALK2 and BMPRII receptor dimer, resulting in stabilization of the BMP receptor complex and, thereby, support of BMP6-Smad signaling ((Benn et al., 2016).
In accordance with VE-cadherin's importance in cell adhesion and multiple signaling pathways, its ablation was lethal at E9.5 due to defects in endothelial cells survival and remodeling. Full knockout of VE-cadherin in mice leads to early embryonic lethality due to severe vascular defects and abnormal somite and heart development (Carmeliet et al., 1999; Gory-Faure et al., 1999). Detailed analysis showed that VE-cadherin is not required for initial vasculogenesis to occur in E8.0 KO embryos. However, at E8.5, mutant vessels were severely affected and although blood vessels formed, they were not maintained (Gory-Faure et al., 1999). Endothelial and endocardial cells dissociated and further detachment between endothelial cells was observed during sprouting angiogenesis. By E9.5, mutant vessels regressed and collapsed, resulting in complete cardiovascular failure that led to embryo proper necrosis.
These observations, as well as a large number of other studies, suggest that during development, VE-cadherin is required to stabilize existing blood vessels as well as to attenuate extracellular proangiogenic signals (reviewed by (Giannotta et al., 2013). Interestingly, abnormal cardiac development was observed in VE-cadherin mutant mice at E8.5, although it is not clear how VE-cadherin AJs affect cardiac differentiation (Carmeliet et al., 1999).
Table 1
VE-cadherin and N-cadherin KO or tissue specific KO in mice
| Gene | Mutant Animals | Phenotype | Refs | |
| N-cadherin | mKO | Lethal at Ε10 |
Developmental defects Cardiovascular heart tube deformation Cardiovascular defects caused by cardiomyocyte inability to aggregate |
(Radice et al., 1997) |
|
mCardiac KO (αMHC) |
Lethal in few months |
Loss of intercalated disc structures. Electrical connectivity defects N-cadherin is not required for cardiomyocytes formation but for proper alignment of myofibrils |
(Kostetskii et al., 2005) |
|
| mEndothelial KO (tie2) | Lethal at the middle of gestation | Severe cardiovascular defects | (Luo and Radice, 2005) | |
| VE-cadherin | mKO | Lethal at Ε9.5 |
Cardiovascular abnormalities Inhibition of primary plexus maturation Defects in endothelial cells survival and remodelling Cardiovascular abnormalities |
(Carmeliet et al., 1999; Gory-Faure et al., 1999) |
VE-cadherin expression during Zebrafish development
VE-cadherin is expressed in Zebrafish, a system that provides powerful tools for dissecting early stages of cardiovascular differentiation. VE-cadherin was found to be expressed before the establishment of cardiovascular system, 12 hpf at the edge of anterior and trunk mesoderm (Larson et al., 2004). Analysis of mutant Zebrafish offered several interesting phenotypes that they have been either confirmed in mammalian models or await further investigation.
A prominent phenotype detected after VE-cadherin knockdown is the formation of supernumerous endothelial cell processes in the intersegmental vessels. Increased sprouting in mutants is caused by activation VEGFR2 signaling, which VE-cadherin antagonize in wild type zebrafish (Abraham et al., 2009; Montero-Balaguer et al., 2009). This phenotype is similar to hypersprouting within the retinal vasculature caused by tamoxifen-induced loss of VE-cadherin function in postnatal mice (Gaengel et al., 2012).
In wild-type embryos, anastomosis of blood vessels in the head is initiated by the formation of a single intercellular junction between filopodia of two opposing tip cells (Paatero et al., 2018). When VE-cadherin function is lost, this adhesion is unstable, and as a result, numerous protrusions form between the tip cells, which however collapse. This demonstrates the importance of VE-cadherin in establishing the first junctional contact necessary for vessels anastomosis. Interestingly, vessels do not seem to be perfused in VE-cadherin mutants, possibly due to cardiac defects (Lenard et al., 2013).
Null mutations in the zebrafish VE-cadherin gene also resulted in impaired junctional remodeling and endothelial cell membrane protrusions formation in mutant embryos. It seems that VE-cadherin not only organizes the cortical actin cytoskeleton via α- and β- catenins anchorage, but also induces actin polymerization in endothelial progenitors by an unknown mechanism (Sauteur et al., 2014). In another study, VE-cadherin expression was linked to a mechanism leading to large caliber vessel aggregation, alignment and finally vascular lumen formation referred to as “lumen ensheathment”. Specifically, in order to form common cardinal veins (CCV), endothelial cells migrate, delaminate in a VE-cadherin-dependent manner and then align around an existing luminal space. VE-cadherin is also important for the directionality of the collective endothelial cells migration to extend the CCV at a later point when circulating blood cells are already passing through (Helker et al., 2013). In addition to vascular defects, zebrafish VE-cadherin knockdown embryos show profoundly impaired cardiac development. Although initial formation of the heart proceeds normally in knockdown embryos, loss of VE-cadherin caused myocardial thinning, abnormal separation of the endocardial and myocardial layers and abnormalities in cardiac looping. Endocardial cell junctions did not form properly, endocardial permeability increased, and the density of the cardiac jelly was reduced. Primitive hematopoiesis, vasculogenesis, and sprouting were unaffected in these mutants (Mitchell et al., 2010). The role of VE-cadherin in this phenotype has not been investigated. It is possible that unknown signaling pathways are impaired, such as shear stress induced pathways, involving VEGF2 receptor or the Hippo pathway. Other possibilities could be the impairment in cell migratory processes and/or the recruitment of progenitor populations due to VE-cadherin knockdown (Haack and Abdelilah-Seyfried, 2016; Duchemin et al., 2019).
VE-cadherin promoter and isolation of cardiovascular progenitors
Although KO studies showed that VE-cadherin plays major roles in the development of cardiovascular vertebrate tissues, there is little information about the nature of VE-cadherin AJs between cardiovascular progenitors before their specification into mature phenotypes. VE-cadherin’s transient expression has been described in the haemogenic endothelium lineage progenitors (Oberlin et al., 2010; Costa et al., 2012; Park et al., 2013; Gritz and Hirschi, 2016). Results from the Zebrafish system led us to the working hypothesis that VE-cadherin could possibly be expressed transiently in cardiac progenitors at some unidentified stage. To this end, we employed a genetic approach to isolate and characterize cardiovascular progenitor cell populations based on the activity pattern of VE-cadherin promoter.
The heart is formed from two distinct progenitor populations, called the first and second heart fields (FHF and SHF respectively), derived from a common ancestor and characterized by the expression of lineage-specific transcription factors (Vincent and Buckingham, 2010; Kelly et al., 2014). FHF progenitors are derived from the anterior splanchnic mesoderm and form the cardiac crescent that subsequently contributes to left ventricle and inflow tract. SHF progenitors are derived from the pharyngeal mesoderm and contribute mainly to right ventricle and the outflow tract (OFT) (Garry and Olson, 2006). Particularly, Isl1 is hallmark of SHF emergence and Isl1+ cells differentiate further to cardiomyocytes, endothelial, endocardial, and smooth muscle cells (Moretti et al., 2006).
Wnt signaling is pivotal for progenitor cell proliferation in a variety of tissues, such as skeletal, neuronal, hemopoietic and cardiac muscle. Its role during cardiac differentiation has been studied in detail and its activation was found to be essential for proper cardiac specification, progenitor expansion and myocardial growth. Specifically, activation of Wnt/β-catenin at the emergence of Isl1+ cells resulted in the clonal expansion of these cardiovascular progenitors. At a later stage, when Wnt/β-catenin is downregulated, Isl1+ cells differentiated to cardiac and endothelial cells. Interestingly, it was reported that N-cadherin AJs prevent the premature differentiation of anterior heart field progenitors, a subset of the SHF population, by modulating b-catenin’s stability and nuclear translocation (Soh et al., 2014).
Since VE-cadherin is considered endothelial specific, its promoter has been used widely for tissue-specific expression in mouse models. A number of VE-cadherin promoter fragments have been isolated and its activity characterized (Gory et al., 1999; Hisatsune et al., 2005; Prandini et al., 2005). They show activity in the yolk sac first, at E7.5, and in the embryo proper by E8.5. A 2.5kb region upstream of the mouse Cdh5 transcriptional initiation has been used to drive Cre recombinase in various strains of Tg(Cdh5-cre) mice (Alva et al., 2006) and it represents one of the most popular endothelial specific expression vehicles (Payne et al., 2018; Kilani et al., 2019)
We isolated a 2.510kb mouse VE-cadherin promoter region (-2486, + 24 in relation to transcription initiation) (Pvec) (Fig. 1A) and generated Pvec-EGFP modified mouse ESC cells for differentiation experiments (Fig. 1B). The activation of Pvec was studied during ESCs in vitro differentiation by monitoring EGFP expression. Pvec was faithfully activated in endothelial cells and more than 90% of EGFP + cells were also VE-cadherin+ at D8 (Fig. 1C). Activation of Pvec was initiated at approximately D4 of mouse ES differentiation (our unpublished data). This prompted us to isolate the Pvec+ cells early mesodermal population by puromycin selection. FACS analysis for EGFP+ cells during ESCs differentiation in selection medium showed that they were 26% at d4.5, increased to 40% at d6, 60% at d7 and d8 of the total cells (Maltabe et al., 2016). EGFP + selected cells co-expressed, in addition to VE-cadherin and PECAM-1, cardiac developmental stage-specific markers Isl1, GATA4 at D7 (Fig. 2).
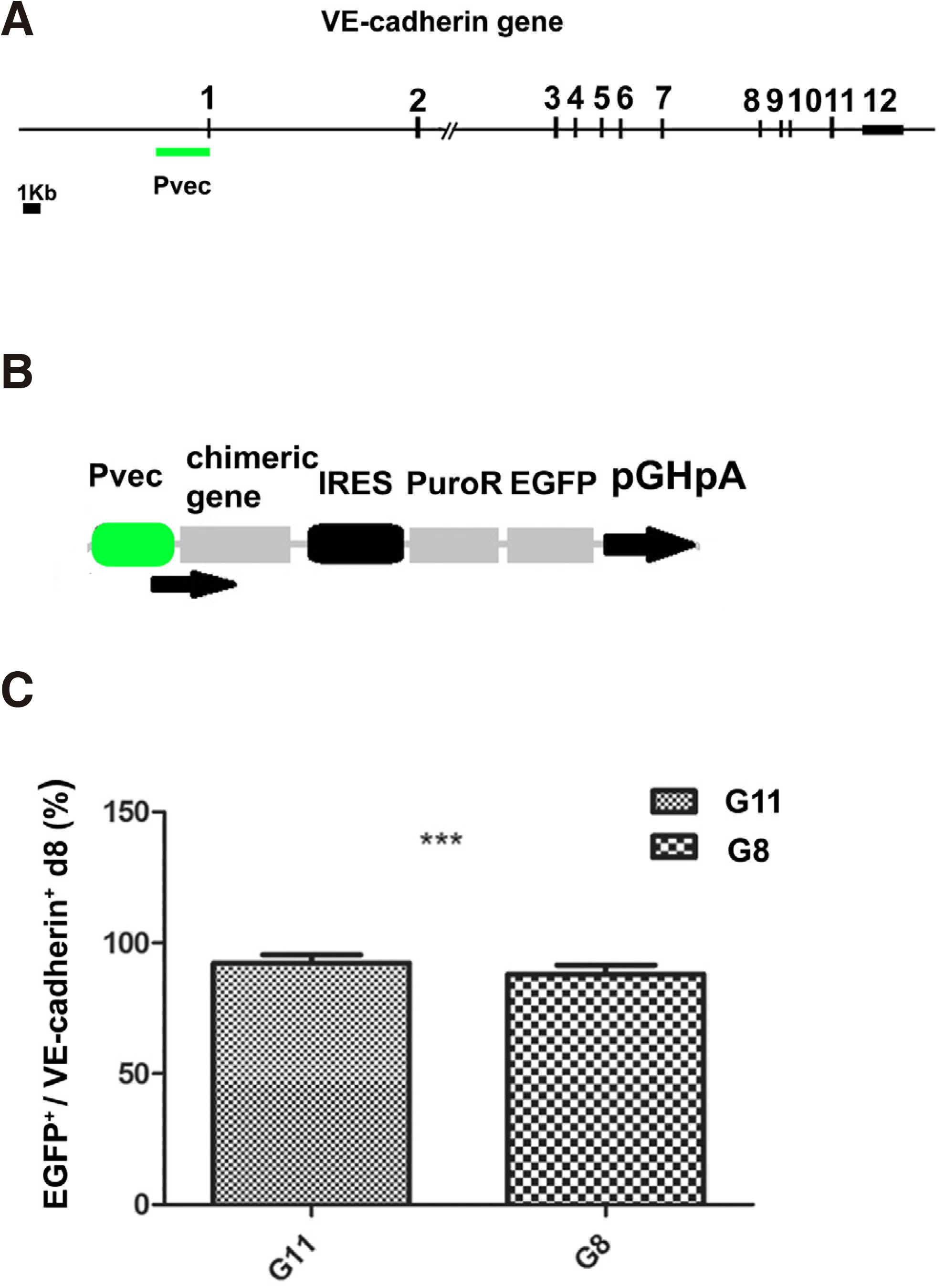
Fig. 1. Generation and characterization of Pvec genetically modified ES clones
(A) Schematic representation of the location of the 12 exons and the VE-cadherin/Cdh5 mouse promoter. (B) Schematic representation of episomal construct pPvec-EGFP, used for generation of ES clones. (C) Statistical analysis of the percentage of VE-cadherin+ cells co-expressing EGFP at d8 from three independent experiments. ***P< 0.001. (from Maltabe et al., 2016).
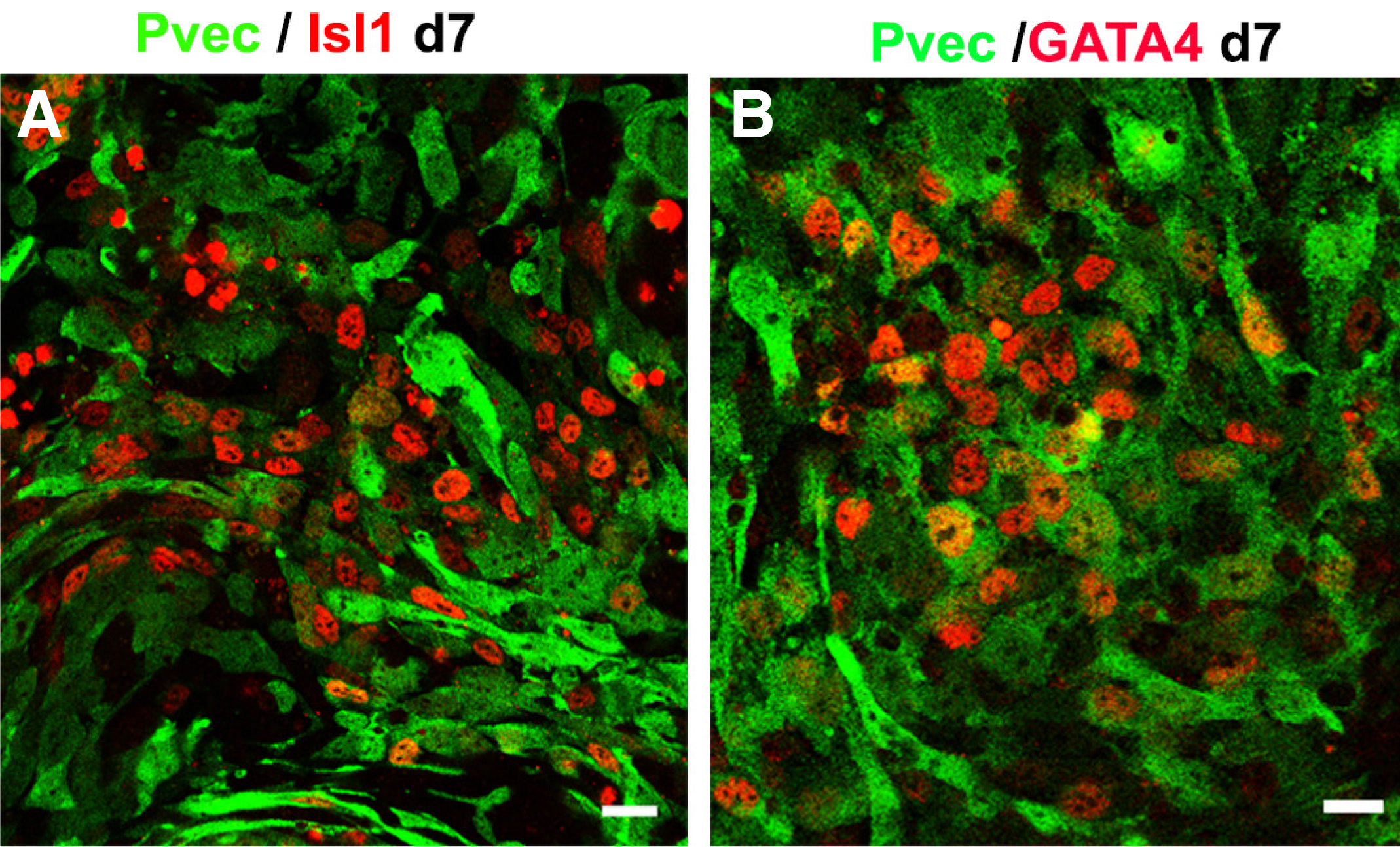
Fig. 2. Pvec+/EGFP+ selected cells co-express Isl1 (red) (A) and GATA4 (red) (B) at d7.
Scale bar 20μm. (From Maltabe et al., 2016). EGFP, enhanced green fluorescent protein.
Pvec+ cells were expanded by addition of SB-216763, a GSK3 inhibitor that induces the Wnt/β-catenin pathway (Fig. 3A, see (Maltabe et al., 2016) for details). As a result, the percentage of Isl1+ cells were found to exceed 60% of total cells at d12 (Fig. 3 B,C). These cells were propagated for at least eight passages up to 30 days and multiple freeze and thaw cycles. Afterwards, their growth declined substantially. (Fig. 3D). Pvec+/Isl1+ cells self-renew evident by co-expression of cardiac progenitor markers GATA4 and Mef2c (Fig. 3 A-C). Further cardiac differentiation was inhibited as indicated by the absence of MLC2v, MLC2a and cTnT expression (Maltabe et al 2016). In addition to cardiac, proliferation of endothelial cells was achieved in Pvec+ selected cells, evident by extensive VE-cadherin-mediated AJs and PECAM-1 junctions (Fig. 4 D-F). Endothelial cells also did not differentiate further, as indicated by the absence of markers of mature endothelium like vWF (Maltabe et al 2016). This finding that implies a positive role of Wnt signaling in endothelial progenitor propagation. Thus, under these culture conditions, a cardiac and endothelial dual-progenitor population able to self-renew was isolated (called CEDPs).
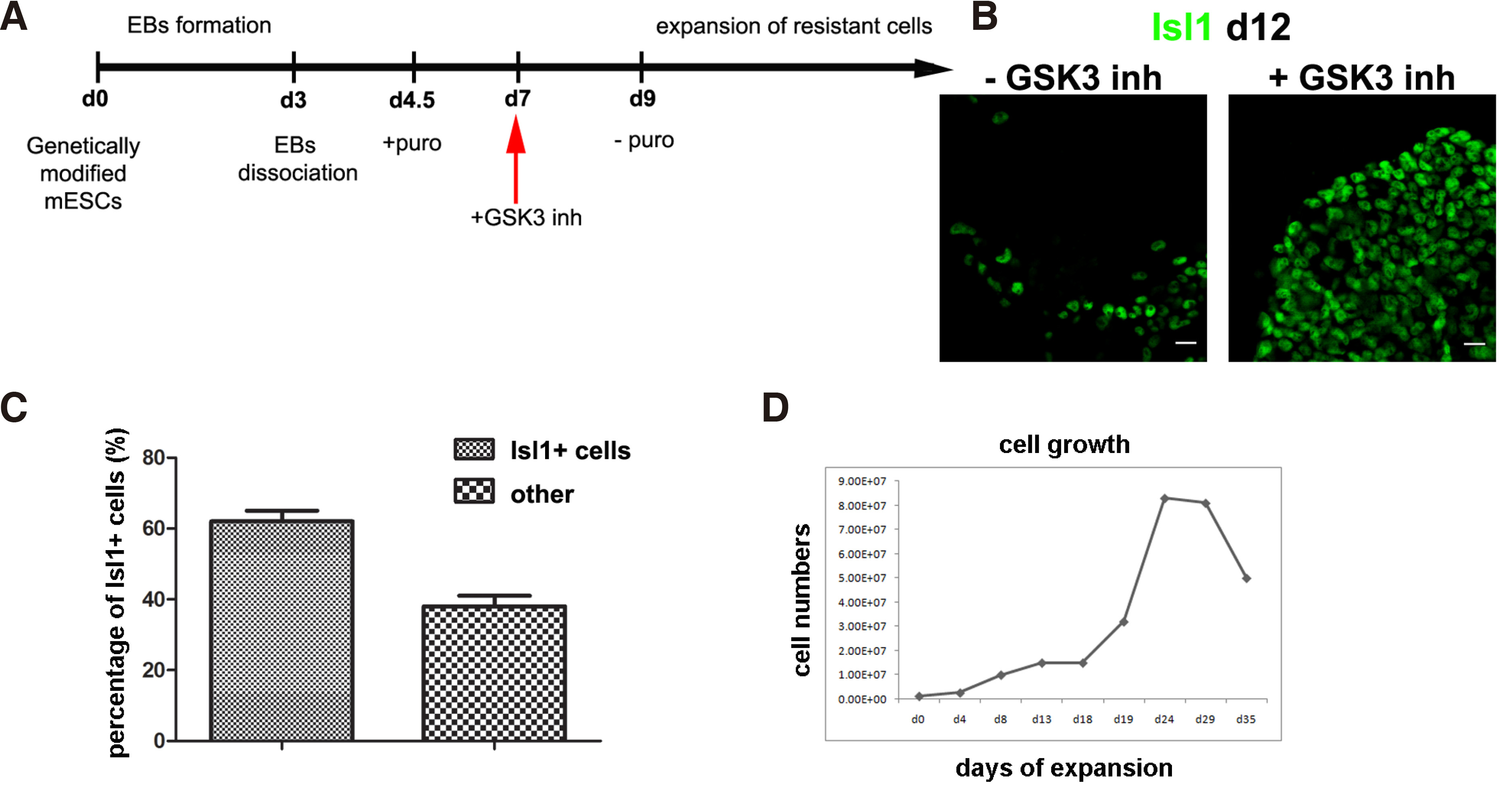
Fig. 3. Cell culture conditions for Expansion of Pvec+ cells.
(A-B) Schematic representation of Pvec+ cell expansion strategy. (B) Wnt/ β-catenin activation induce propagation of Pvec+ cells expressing Isl1 at d12. (C) Pvec+/Isl1+ cells percentage in SB-216763 from three independent experiments. (D) Pvec+ cell growth curve. The doubling -time of Pvec + cells in the presence of SB-216763 calculated after growth curve generation between day 1 to day 28 was ~4.5 days. Scale bar 20μm.
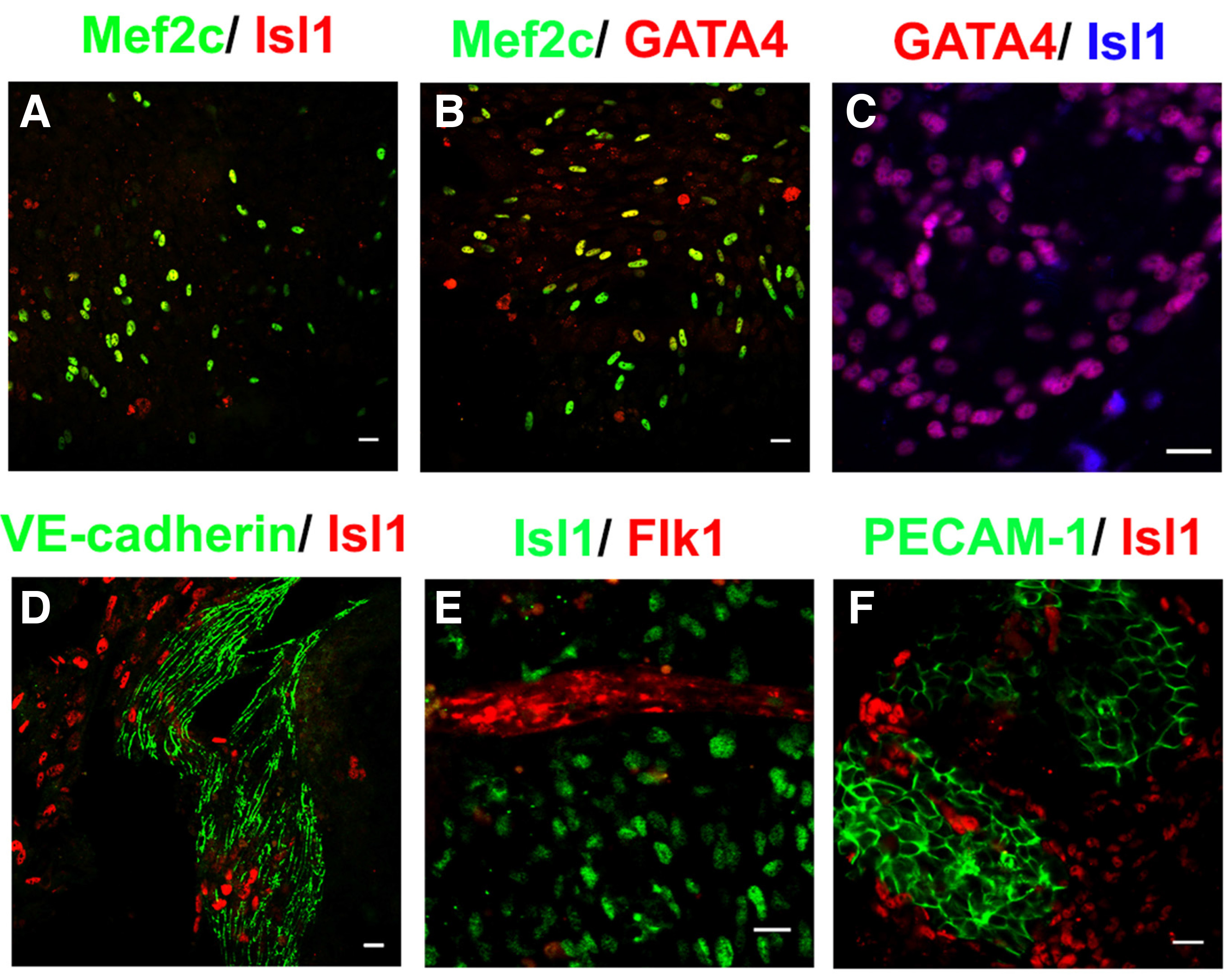
Fig. 4. Characterization of endothelial and cardiac progenitor populations in CEDPs.
(A-F) Propagated cells consist of cell populations expressing markers of cardiac (Isl1, Mef2C, GATA4) and/or endothelial (Flk-1, PECAM-1, VE-cadherin) progenitors. Scale bar 20μm. (from Maltabe et al., 2016).
Survival and differentiation of CEDPs after transplantation in vivo
Transplantation of CEDPs in the left ventricle (LV) of immunosuppressed rats showed that CEDPs survived and differentiated in vivo for at least 14 days (Fig. 5 A,B,D). Transplanted CEDPs differentiated to cardiomyocytes in vivo shown by MLC2v expression, a cardiac myocytes marker not expressed in CEDPs. Differentiation of CEDPs was first detected 3 days after transplantation, shown by MLC2v expression on the 3rd, 7th and 14th day post-transplantation (Fig. 5A). CEDP-derived cells were discriminated from endogenous cardiac cells based on Isl1 expression present only in CEDPs and their progeny, but not in the LV of the adult heart. Quantification of Isl1+ cells that survived seven days post-transplantation dissected LVs showed that surviving Isl1+ cells represent ~22% of the initially injected cells (Fig. 5C), considering that Isl1+ cells percentage is ~60% of CEDPs (Fig. 1C).
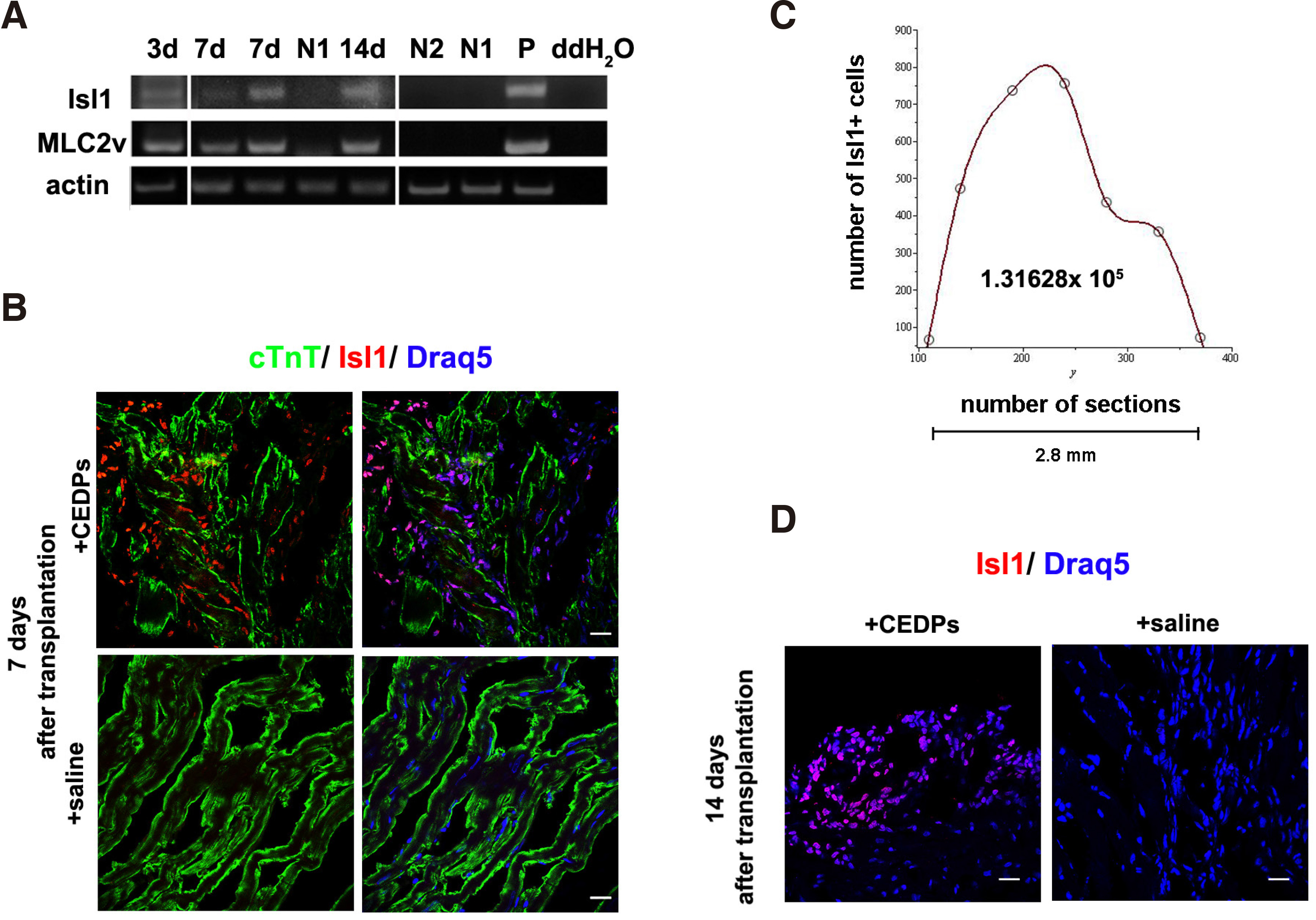
Fig. 5. Survival and differentiation of CEDPs after transplantation in adult rat heart.
(A) Isl1 and MLC2v expression after 3, 7 and 14 days of transplantation by RT-PCR analysis. mRNA from saline-injected immunosuppressed (N1) or untreated (N2) LV of adult rats were used as negative controls and mRNA from E12 mouse embryos (P) as positive control. (B, D) The LV areas from CEDPs or saline recipient rats were dissected and examined after 7 and 14 days by immunocytochemical for survival and differentiation of CEDPs. Immunostaining of frozen sections demonstrated in all cases that Isl1+ cell populations appeared specifically in CEDPs-, but not in saline-injected animals. (C) All Isl1+ cells from two non-sequential sections were photographed and counted with Fiji cell-counter. Draq5 used for DNA staining. The total Isl1+ cell number found in 2.8mm tissue was calculated after Spline interpolation. (Scale bar 20μm. From (Maltabe et al., 2016).
The endothelial specific activity of the Pvec fragment of mouse VE-cadherin promoter has been demonstrated in transgenic animals and during ESCs differentiation. It has been further utilized for endothelial lineage tracing, endothelial specific knock-out mutant mice generation and isolation of ES-derived endothelial cells. However, transient activation of Pvec was also found in hemogenic endothelium, a progenitor population that differentiated to endothelial and definitive hemopoietic cells lineages (Chen et al., 2009). This demonstrates that precise timely evaluation of Pvec activity is crucial for selection experiments. In our laboratory, we have studied in detail the Pvec activity pattern during ESC differentiation. Surprisingly, perhaps, we found that it was transiently activated in a subset of Isl1+ multipotent cardiovascular progenitors between days 4-5; based on this finding, we selected cells surviving at this time-window. This genetically based approach resulted in isolation of the Pvec+ cell population with specific endothelial- and cardiac- progenitor phenotypes, an indication of specific selection.
In conclusion, based on VE-cadherin’s promoter activity pattern during early differentiation, a novel cardiovascular progenitor cell population was isolated and characterized, which has the capacity to self-renew and further differentiate to endothelial, cardiac, and smooth muscle cells in vitro and in vivo.
These cells can be used in a cell-based therapy for myocardial infarct and in drug screening.
Acknowledgements
This research is co-financed by Greece and the European Union (European Social Fund- ESF) through the Operational Programme «Human Resources Development, Education and Lifelong Learning» in the context of the project “Reinforcement of Postdoctoral Researchers - 2nd Cycle” (MIS-5033021), implemented by the State Scholarships Foundation (ΙΚΥ). The project is co-financed by Greece and the European Union - European Regional Development Fund (ERDF) under the Operational Program "Competitiveness Entrepreneurship Innovation" (EPAnEK), NSRF 2014-2020 (MIS 5047236).
Abbreviations
AJs, Adherens Junctions ; CEDPs, Cardiac and Endothelial dual-progenitor ; CCV, Common cardinal veins ; EGFP, enhanced green fluorescent protein ; ESCs, Embryonic Stem Cells ; FHF, First Heart Field ; KO, Knockout ; LV, Left ventricle ; MAPK, Mitogen-activated protein kinase ; OFT, Outflow tract ; SHF, Second Heart Field ; TGF-β, Transforming growth factor-beta ; VEGFR2, Vascular-endothelial growth factor receptor 2 ; Pvec, VE-cadherin promoter ;References
Abraham S., Yeo M., Montero-Balaguer M., Paterson H., Dejana E., Marshall C. J., Mavria G. (2009). VE-Cadherin-Mediated Cell-Cell Interaction Suppresses Sprouting via Signaling to MLC2 Phosphorylation. Current Biology 19: 668-674.
Acloque H., Adams M. S., Fishwick K., Bronner-Fraser M., Nieto M. A. (2009). Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. Journal of Clinical Investigation 119: 1438-1449.
Alva J. A., Zovein A. C., Monvoisin A., Murphy T., Salazar A., Harvey N. L., Carmeliet P., Iruela-Arispe M. L. (2006). VE-Cadherin-Cre-recombinase transgenic mouse: A tool for lineage analysis and gene deletion in endothelial cells. Developmental Dynamics 235: 759-767.
Benn A., Bredow C., Casanova I., Vukičević S., Knaus P. (2016). VE-Cadherin facilitates BMP-induced endothelial cell permeability and signaling. Journal of Cell Science 129: 206-218.
Carmeliet P., Lampugnani M.G., Moons L., Breviario F., Compernolle V., Bono F., Balconi G., Spagnuolo R., Oosthuyse B., Dewerchin M., Zanetti A., Angellilo A., Mattot V., Nuyens D., Lutgens E., Clotman F., de Ruiter M. C., Gittenberger-de Groot A., Poelmann R., Lupu F., Herbert J.M., Collen D., Dejana E. (1999). Targeted Deficiency or Cytosolic Truncation of the VE-cadherin Gene in Mice Impairs VEGF-Mediated Endothelial Survival and Angiogenesis. Cell 98: 147-157.
Chen M. J., Yokomizo T., Zeigler B. M., Dzierzak E., Speck N. A. (2009). Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 457: 887-891.
Choi H.J., Kwon Y.G. (2015). Roles of YAP in mediating endothelial cell junctional stability and vascular remodeling. BMB Reports 48: 429-430.
Conway D. E., Breckenridge M. T., Hinde E., Gratton E., Chen C. S., Schwartz M. A. (2013). Fluid Shear Stress on Endothelial Cells Modulates Mechanical Tension across VE-Cadherin and PECAM-1. Current Biology 23: 1024-1030.
Costa G., Mazan A., Gandillet A., Pearson S., Lacaud G., Kouskoff V. (2012). SOX7 regulates the expression of VE-cadherin in the haemogenic endothelium at the onset of haematopoietic development. Development 139: 1587-1598.
Dejana E., Vestweber D. (2013). The Role of VE-Cadherin in Vascular Morphogenesis and Permeability Control. In The Molecular Biology of Cadherins. Elsevier.
Duchemin A.L., Vignes H., Vermot J., Chow R. (2019). Mechanotransduction in cardiovascular morphogenesis and tissue engineering. Current Opinion in Genetics & Development 57: 106-116.
Gaengel K., Niaudet C., Hagikura K., Laviña B., Muhl L., Hofmann J. J., Ebarasi L., Nyström S., Rymo S., Chen L. L., Pang M.F., Jin Y., Raschperger E., Roswall P., Schulte D., Benedito R., Larsson J., Hellström M., Fuxe J., Uhlén P., Adams R., Jakobsson L., Majumdar A., Vestweber D., Uv A., Betsholtz C. (2012). The Sphingosine-1-Phosphate Receptor S1PR1 Restricts Sprouting Angiogenesis by Regulating the Interplay between VE-Cadherin and VEGFR2. Developmental Cell 23: 587-599.
Garry D. J., Olson E. N. (2006). A Common Progenitor at the Heart of Development. Cell 127: 1101-1104.
Giampietro C., Disanza A., Bravi L., Barrios-Rodiles M., Corada M., Frittoli E., Savorani C., Lampugnani M. G., Boggetti B., Niessen C., Wrana J. L., Scita G., Dejana E. (2015). The actin-binding protein EPS8 binds VE-cadherin and modulates YAP localization and signaling. Journal of Cell Biology 211: 1177-1192.
Giannotta M., Trani M., Dejana E. (2013). VE-Cadherin and Endothelial Adherens Junctions: Active Guardians of Vascular Integrity. Developmental Cell 26: 441-454.
Gory-Faure S., Prandini M.H., Pointu H., Roullot V., Pignot-Paintrand I., Vernet M., Huber P. (1999). Role of vascular endothelial-cadherin in vascular morphogenesis. Development 126: 2093-2102.
Gory S., Vernet M., Laurent M., Dejana E., Dalmon J., Huber P. (1999). The Vascular Endothelial-Cadherin Promoter Directs Endothelial-Specific Expression in Transgenic Mice. Blood 93: 184-192.
Lampugnani M. G., Zanetti A., Corada M., Takahashi T., Balconi G., Breviario F., Orsenigo F., Cattelino A., Kemler R., Daniel T. O., Dejana E. (2003). Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, β-catenin, and the phosphatase DEP-1/CD148. Journal of Cell Biology 161: 793-804.
Gritz E., Hirschi K. K. (2016). Specification and function of hemogenic endothelium during embryogenesis. Cellular and Molecular Life Sciences 73: 1547-1567.
Haack T., Abdelilah-Seyfried S. (2016). The force within: endocardial development, mechanotransduction and signalling during cardiac morphogenesis. Development 143: 373-386.
Halbleib J. M., Nelson W. J., (2006). Cadherins in development: cell adhesion, sorting, and tissue morphogenesis. Genes & Development 20: 3199-3214.
Harris T. J. C., Tepass U. (2010). Adherens junctions: from molecules to morphogenesis. Nature Reviews Molecular Cell Biology 11: 502-514.
Helker C. S. M., Schuermann A., Karpanen T., Zeuschner D., Belting H.G., Affolter M., Schulte-Merker S., Herzog W. (2013). The zebrafish common cardinal veins develop by a novel mechanism: lumen ensheathment. Development 140: 2776-2786.
Hisatsune H. (2005). High level of endothelial cell-specific gene expression by a combination of the 5' flanking region and the 5' half of the first intron of the VE-cadherin gene. Blood 105: 4657-4663.
Hulpiau P., van Roy F. (2009). Molecular evolution of the cadherin superfamily. The International Journal of Biochemistry & Cell Biology 41: 349-369.
Iden S., Rehder D., August B., Suzuki A., Wolburg‐Buchholz K., Wolburg H., Ohno S., Behrens J., Vestweber D., Ebnet K. (2006). A distinct PAR complex associates physically with VE‐cadherin in vertebrate endothelial cells. EMBO reports 7: 1239-1246.
Kelly R. G., Buckingham M. E., Moorman A. F. (2014). Heart Fields and Cardiac Morphogenesis. Cold Spring Harbor Perspectives in Medicine 4: a015750-a015750.
Kilani B., Gourdou‐Latyszenok V., Guy A., Bats M.L., Peghaire C., Parrens M., Renault M.A., Duplàa C., Villeval J.L., Rautou P.E., Couffinhal T., James C. (2019). Comparison of endothelial promoter efficiency and specificity in mice reveals a subset of Pdgfb‐positive hematopoietic cells. Journal of Thrombosis and Haemostasis 17: 827-840.
Klezovitch O., Vasioukhin V. (2015). Cadherin signaling: keeping cells in touch. F1000Research 4: 550.
Komarova Y. A., Kruse K., Mehta D., Malik A. B. (2017). Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circulation Research 120: 179-206.
Kostetskii I., Li J., Xiong Y., Zhou R., Ferrari V. A., Patel V. V., Molkentin J. D., Radice G. L. (2005). Induced Deletion of the N-Cadherin Gene in the Heart Leads to Dissolution of the Intercalated Disc Structure . Circulation Research 96: 346-354.
Lampugnani M. G., Orsenigo F., Gagliani M. C., Tacchetti C., Dejana E. (2006). Vascular endothelial cadherin controls VEGFR-2 internalization and signaling from intracellular compartments. Journal of Cell Biology 174: 593-604.
Larson J. D., Wadman S. A., Chen E., Kerley L., Clark K. J., Eide M., Lippert S., Nasevicius A., Ekker S. C., Hackett P. B., Essner J. J. (2004). Expression ofVE-cadherin in zebrafish embryos: A new tool to evaluate vascular development. Developmental Dynamics 231: 204-213.
Lenard A., Ellertsdottir E., Herwig L., Krudewig A., Sauteur L., Belting H.G., Affolter M. (2013). In Vivo Analysis Reveals a Highly Stereotypic Morphogenetic Pathway of Vascular Anastomosis. Developmental Cell 25: 492-506.
Luo Y., Radice G. L. (2005). N-cadherin acts upstream of VE-cadherin in controlling vascular morphogenesis. Journal of Cell Biology 169: 29-34.
Maltabe V. A., Barka E., Kontonika M., Florou D., Kouvara-Pritsouli M., Roumpi M., Agathopoulos S., Kolettis T. M., Kouklis P. (2016). Isolation of an ES-Derived Cardiovascular Multipotent Cell Population Based on VE-Cadherin Promoter Activity. Stem Cells International 2016: 1-14.
Mitchell I. C., Brown T. S., Terada L. S., Amatruda J. F., Nwariaku F. E. (2010). Effect of Vascular Cadherin Knockdown on Zebrafish Vasculature during Development. PLoS ONE 5: e8807.
Montero-Balaguer M., Swirsding K., Orsenigo F., Cotelli F., Mione M., Dejana E. (2009). Stable Vascular Connections and Remodeling Require Full Expression of VE-Cadherin in Zebrafish Embryos. PLoS ONE 4: e5772.
Moretti A., Caron L., Nakano A., Lam J. T., Bernshausen A., Chen Y., Qyang Y., Bu L., Sasaki M., Martin-Puig S., Sun Y., Evans S. M., Laugwitz K.L., Chien K. R. (2006). Multipotent Embryonic Isl1+ Progenitor Cells Lead to Cardiac, Smooth Muscle, and Endothelial Cell Diversification. Cell 127: 1151-1165.
Niessen C. M., Leckband D., Yap A. S. (2011). Tissue Organization by Cadherin Adhesion Molecules: Dynamic Molecular and Cellular Mechanisms of Morphogenetic Regulation. Physiological Reviews 91: 691-731.
Oberlin E., Fleury M., Clay D., Petit-Cocault L., Candelier J.J., Mennesson B., Jaffredo T., Souyri M. (2010). VE-cadherin expression allows identification of a new class of hematopoietic stem cells within human embryonic liver. Blood 116: 4444-4455.
Paatero I., Sauteur L., Lee M., Lagendijk A. K., Heutschi D., Wiesner C., Guzmán C., Bieli D., Hogan B. M., Affolter M., Belting H.G. (2018). Junction-based lamellipodia drive endothelial cell rearrangements in vivo via a VE-cadherin-F-actin based oscillatory cell-cell interaction. Nature Communications 9: 3545.
Park T. S., Zimmerlin L., Zambidis E. T. (2013). Efficient and simultaneous generation of hematopoietic and vascular progenitors from human induced pluripotent stem cells. Cytometry Part A 83A: 114-126.
Payne S., De Val S., Neal A. (2018). Endothelial-Specific Cre Mouse Models. Arteriosclerosis, Thrombosis, and Vascular Biology 38: 2550-2561.
Prandini M.H., Dreher I., Bouillot S., Benkerri S., Moll T., Huber P. (2005). The human VE-cadherin promoter is subjected to organ-specific regulation and is activated in tumour angiogenesis. Oncogene 24: 2992-3001.
Radice G. L., Rayburn H., Matsunami H., Knudsen K. A., Takeichi M., Hynes R. O. (1997). Developmental Defects in Mouse Embryos Lacking N-Cadherin. Developmental Biology 181: 64-78.
Rudini N., Felici A., Giampietro C., Lampugnani M.G., Corada M., Swirsding K., Garrè M., Liebner S., Letarte M., ten Dijke P., Dejana E. (2008). VE-cadherin is a critical endothelial regulator of TGF-β signalling. The EMBO Journal 27: 993-1004.
Sauteur L., Krudewig A., Herwig L., Ehrenfeuchter N., Lenard A., Affolter M., Belting H.G. (2014). Cdh5/VE-cadherin Promotes Endothelial Cell Interface Elongation via Cortical Actin Polymerization during Angiogenic Sprouting. Cell Reports 9: 504-513.
Soh B.S., Buac K., Xu H., Li E., Ng S.Y., Wu H., Chmielowiec J., Jiang X., Bu L., Li R. A., Cowan C., Chien K. R. (2014). N-cadherin prevents the premature differentiation of anterior heart field progenitors in the pharyngeal mesodermal microenvironment. Cell Research 24: 1420-1432.
Stepniak E., Radice G. L., Vasioukhin V. (2009). Adhesive and Signaling Functions of Cadherins and Catenins in Vertebrate Development. Cold Spring Harbor Perspectives in Biology 1: a002949-a002949.
Tzima E., Irani-Tehrani M., Kiosses W. B., Dejana E., Schultz D. A., Engelhardt B., Cao G., DeLisser H., Schwartz M. A. (2005). A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437: 426-431.
Vincent S. D., Buckingham M. E. (2010). How to Make a Heart: The Origin and Regulation of Cardiac Progenitor Cells. In Organogenesis in Development. Elsevier.
Wheelock M. J., Shintani Y., Maeda M., Fukumoto Y., Johnson K. R. (2008). Cadherin switching. Journal of Cell Science 121: 727-735.